Following the previous bug report on fix_vector.cpp, I restarted a simulation with the patched version of LAMMPS (22 Jul 2025 - Update 4) and found that the equilibrium state of an equilibrated sample quickly moved to a different state. See the evolution of the density:
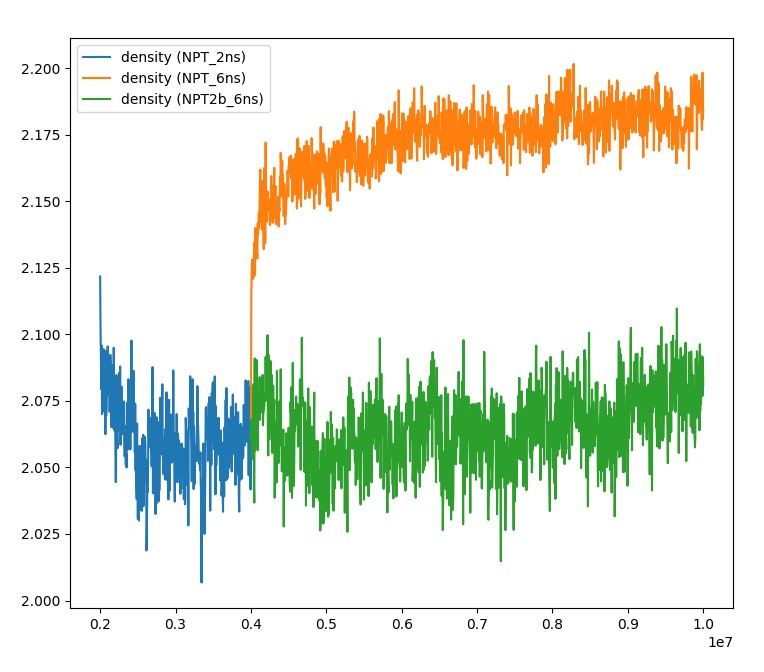
The orange line is the trajectory computed with the patched version (update 4), while the green and blue lines are computed with LAMMPS (22 Jul 2025).
The orange and green simulations come from identical input files and use the same restart file. They are just executed on two different versions of LAMMPS (22Jul25 and 22Jul25_update4). The two LAMMPS executables are compiled with the same compiler and the simulations carried out on the same cluster. I also tested the published 22Jul25_update4 without patch, and it gives the same results as the orange trajectory, so the difference is not due to the patch discussed in the previous post.
I am reporting this behaviour because this is a pretty vanilla setup with very established potential functions, so I wasn’t expecting any dramatic difference between two very recent LAMMPS versions (as I wouldn’t doubt simulations carried out 20 years ago with LAMMPS using the same potentials). Anyway, when I compare the starting point with the two executables, this is what I see:
@@ -1,3 +1,3 @@
-LAMMPS (22 Jul 2025 - Update 4)
+LAMMPS (22 Jul 2025)
using 1 OpenMP thread(s) per MPI task
package intel 1
@@ -175,7 +175,7 @@
pair build: half/bin/newton/tri/intel
stencil: half/bin/3d/tri/intel
bin: intel
-Per MPI rank memory allocation (min/avg/max) = 8.496 | 8.583 | 8.774 Mbytes
+Per MPI rank memory allocation (min/avg/max) = 8.26 | 8.346 | 8.531 Mbytes
Step TotEng E_vdwl E_coul E_long E_bond E_angle E_dihed E_impro KinEng PotEng Temp Press Volume Density v_msqdis v_diff CPU
- 4000000 -189880.6 16398.021 -114802.43 -93860.196 639.61688 167.74865 0 0 1576.64 -191457.24 298.99946 -6681.2718 21698.441 2.060053 0 3.3333333e+15 0
- 4005000 -189929.55 16872.598 -115377.21 -93854.541 655.80918 165.51629 0 0 1608.2721 -191537.82 304.99829 -466.14381 21136.17 2.1148551 0.74800541 2.4933514e-05 6.8582337
+ 4000000 -190009.31 16398.021 -114802.42 -93860.196 520.80711 157.84788 0 0 1576.64 -191585.95 298.99946 -6681.2697 21698.441 2.060053 0 3.3333333e+15 0
+ 4005000 -190052.61 16498.483 -114954.62 -93856.802 538.06563 151.57617 0 0 1570.6846 -191623.3 297.87006 -5220.7239 21617.197 2.0677953 0.48500297 1.6166766e-05 3.1489684
The takeaways:
- The pairwise energy contributions (E_vdwl, E_coul, E_long) are the same. The virial must also be the same, as the pressure is idential at step 4000000.
- The E_bond and E_angle are different.
- The density in 22Jul25_update4 increases quite rapidly.
I am not sure what drives this change in the two trajectories, but it’s not caused by the patch to fix vector. If you want to reproduce this simulation, please use the input deck in this post.
Thank you,
Otello