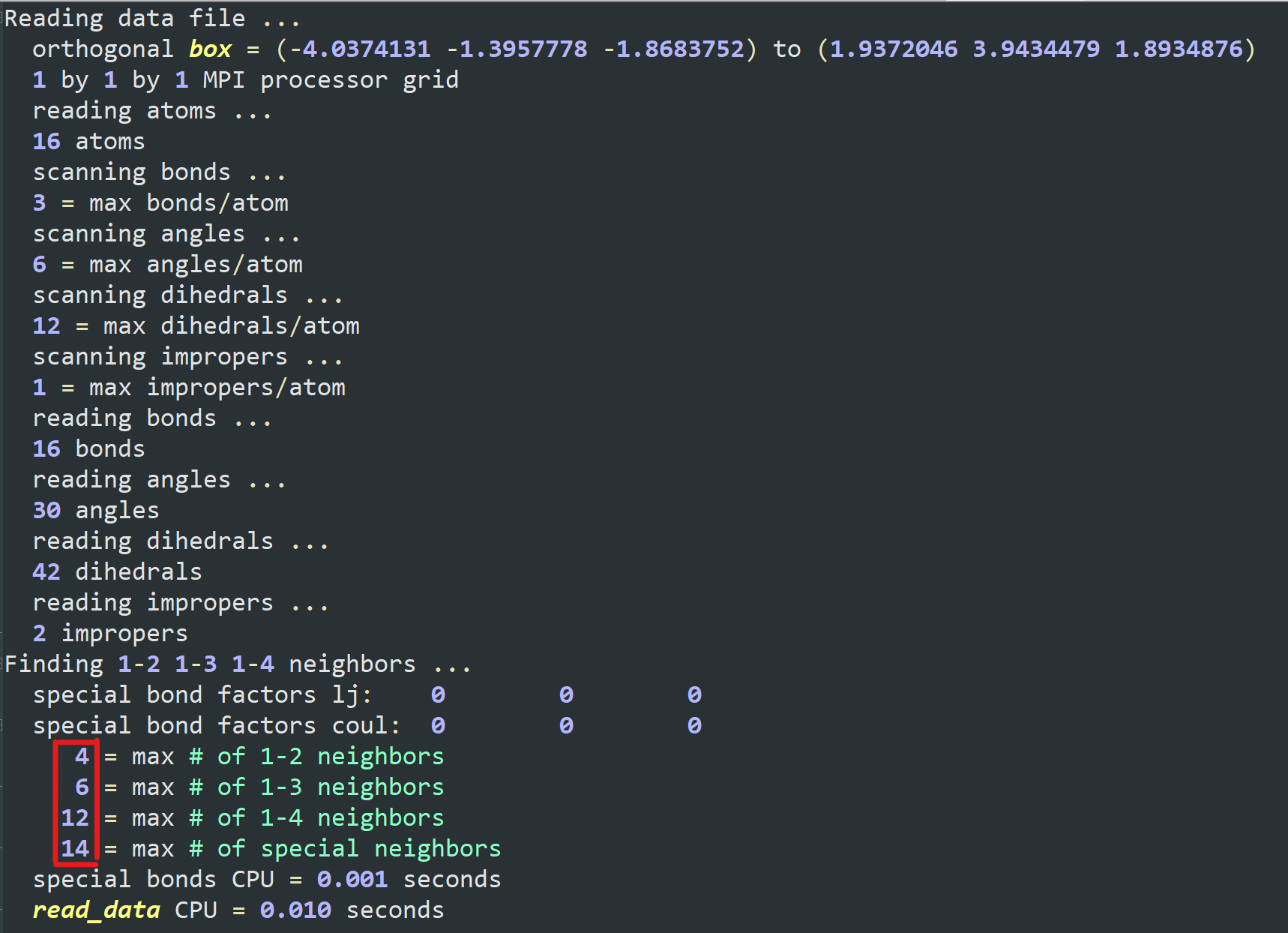
I want to add some PIP molecules to the system. The pip.data is established in materials studio converting by mis2lmp.exe. And I wrote the PIP.text according to the pip.data. However, I got trouble in the create_box command. I don’t konw the value of the “extra/bond/per/atom”, the value of the “extra/angle/per/atom” and the value of the “extra/dihedral/per/atom” refer to what. Every time I run the program, the error is “Molecule auto special bond generation overflow.” I think maybe I used create_box command wrong, but I don’t konw how to correct it. I hope someone can help me. Thanks a lot.
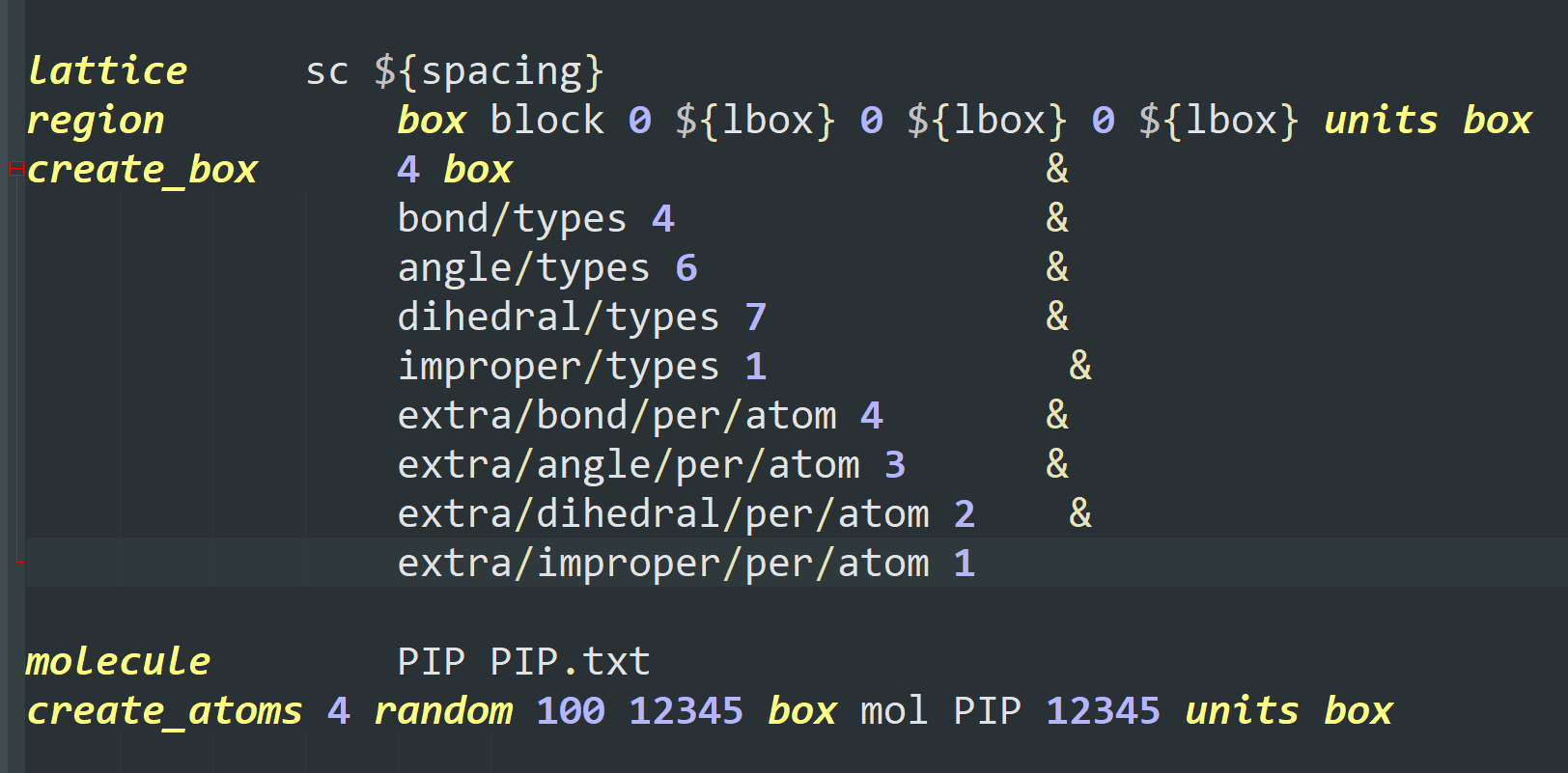
Following is the pip.data.
LAMMPS data file. msi2lmp v3.9.9 / 05 Nov 2018 / CGCMM for .\pip
16 atoms
16 bonds
30 angles
42 dihedrals
2 impropers
4 atom types
4 bond types
6 angle types
7 dihedral types
1 improper types
-4.037413097 1.937204622 xlo xhi
-1.395777808 3.943447890 ylo yhi
-1.868375189 1.893487555 zlo zhi
Masses
1 14.006700 # n3
2 12.011150 # c2
3 1.007970 # hn
4 1.007970 # h
Pair Coeffs # lj/cut/coul/long
1 0.1669999743 3.5012320066 # n3
2 0.0389999952 3.8754094636 # c2
3 0.0000000000 0.0000000000 # hn
4 0.0380000011 2.4499714540 # h
Bond Coeffs # harmonic
1 356.5988 1.4700 # n3-c2
2 457.4592 1.0260 # n3-hn
3 322.7158 1.5260 # c2-c2
4 340.6175 1.1050 # c2-h
Angle Coeffs # harmonic
1 86.3000 112.0000 # c2-n3-c2
2 41.6000 110.0000 # c2-n3-hn
3 50.0000 109.5000 # n3-c2-c2
4 44.4000 110.0000 # c2-c2-h
5 57.3000 109.5000 # n3-c2-h
6 39.5000 106.4000 # h-c2-h
Dihedral Coeffs # harmonic
1 0.1333 1 3 # c2-n3-c2-c2
2 0.1333 1 3 # c2-n3-c2-h
3 0.1333 1 3 # hn-n3-c2-c2
4 0.1333 1 3 # hn-n3-c2-h
5 0.1581 1 3 # n3-c2-c2-n3
6 0.1581 1 3 # n3-c2-c2-h
7 0.1581 1 3 # h-c2-c2-h
Improper Coeffs # cvff
1 0.0000 0 0 # c2-n3-c2-hn
Atoms # full
1 1 1 0.000000 -2.474156782 1.268292122 0.330146493 0 0 0 # n3
2 1 2 0.000000 -1.778245099 0.031909754 -0.186352597 0 0 0 # c2
3 1 2 0.000000 -0.303136828 0.050902422 0.255088784 0 0 0 # c2
4 1 1 0.000000 0.373974982 1.278718503 -0.304920390 0 0 0 # n3
5 1 2 0.000000 -0.321884506 2.515264494 0.211388608 0 0 0 # c2
6 1 2 0.000000 -1.797068954 2.496065671 -0.229961299 0 0 0 # c2
7 1 3 0.000000 -3.537413097 1.254597474 0.011740002 0 0 0 # hn
8 1 4 0.000000 -2.289242491 -0.895777808 0.235323721 0 0 0 # h
9 1 4 0.000000 -1.833705760 0.012439858 -1.324812564 0 0 0 # h
10 1 4 0.000000 0.222066675 -0.883201907 -0.133705889 0 0 0 # h
11 1 4 0.000000 -0.247432421 0.072379544 1.393487555 0 0 0 # h
12 1 3 0.000000 1.437204622 1.292451665 0.013544842 0 0 0 # hn
13 1 4 0.000000 0.188030819 3.443447890 -0.210585219 0 0 0 # h
14 1 4 0.000000 -0.266153624 2.534910192 1.349840338 0 0 0 # h
15 1 4 0.000000 -1.852606463 2.474325256 -1.368375189 0 0 0 # h
16 1 4 0.000000 -2.322227579 3.430170544 0.158878506 0 0 0 # h
Bonds
1 1 1 2
2 1 1 6
3 2 1 7
4 3 2 3
5 4 2 8
6 4 2 9
7 1 4 3
8 4 3 10
9 4 3 11
10 1 4 5
11 2 4 12
12 3 5 6
13 4 5 13
14 4 5 14
15 4 6 15
16 4 6 16
Angles
1 1 2 1 6
2 2 2 1 7
3 2 6 1 7
4 3 1 2 3
5 4 3 2 8
6 4 3 2 9
7 5 1 2 8
8 5 1 2 9
9 6 8 2 9
10 3 4 3 2
11 4 2 3 10
12 4 2 3 11
13 5 4 3 10
14 5 4 3 11
15 6 10 3 11
16 1 3 4 5
17 2 3 4 12
18 2 5 4 12
19 3 4 5 6
20 5 4 5 13
21 5 4 5 14
22 4 6 5 13
23 4 6 5 14
24 6 13 5 14
25 3 1 6 5
26 4 5 6 15
27 4 5 6 16
28 5 1 6 15
29 5 1 6 16
30 6 15 6 16
Dihedrals
1 1 6 1 2 3
2 2 6 1 2 8
3 2 6 1 2 9
4 3 7 1 2 3
5 4 7 1 2 8
6 4 7 1 2 9
7 1 2 1 6 5
8 2 2 1 6 15
9 2 2 1 6 16
10 3 7 1 6 5
11 4 7 1 6 15
12 4 7 1 6 16
13 5 1 2 3 4
14 6 1 2 3 10
15 6 1 2 3 11
16 6 4 3 2 8
17 7 8 2 3 10
18 7 8 2 3 11
19 6 4 3 2 9
20 7 9 2 3 10
21 7 9 2 3 11
22 1 5 4 3 2
23 3 12 4 3 2
24 2 5 4 3 10
25 4 12 4 3 10
26 2 5 4 3 11
27 4 12 4 3 11
28 1 3 4 5 6
29 2 3 4 5 13
30 2 3 4 5 14
31 3 12 4 5 6
32 4 12 4 5 13
33 4 12 4 5 14
34 5 4 5 6 1
35 6 4 5 6 15
36 6 4 5 6 16
37 6 1 6 5 13
38 7 13 5 6 15
39 7 13 5 6 16
40 6 1 6 5 14
41 7 14 5 6 15
42 7 14 5 6 16
Impropers
1 1 2 1 6 7
2 1 3 4 5 12
Following is the PIP.txt:
PIP molecule.
16 atoms
16 bonds
30 angles
42 dihedrals
2 impropers
Coords
1 -2.474156782 1.268292122 0.330146493
2 -1.778245099 0.031909754 -0.186352597
3 -0.303136828 0.050902422 0.255088784
4 0.373974982 1.278718503 -0.304920390
5 -0.321884506 2.515264494 0.211388608
6 -1.797068954 2.496065671 -0.229961299
7 -3.537413097 1.254597474 0.011740002
8 -2.289242491 -0.895777808 0.235323721
9 -1.833705760 0.012439858 -1.324812564
10 0.222066675 -0.883201907 -0.133705889
11 -0.247432421 0.072379544 1.393487555
12 1.437204622 1.292451665 0.013544842
13 0.188030819 3.443447890 -0.210585219
14 -0.266153624 2.534910192 1.349840338
15 -1.852606463 2.474325256 -1.368375189
16 -2.322227579 3.430170544 0.158878506
Types
1 1
2 2
3 2
4 1
5 2
6 2
7 3
8 4
9 4
10 4
11 4
12 3
13 4
14 4
15 4
16 4
Charges
1 0.000000
2 0.000000
3 0.000000
4 0.000000
5 0.000000
6 0.000000
7 0.000000
8 0.000000
9 0.000000
10 0.000000
11 0.000000
12 0.000000
13 0.000000
14 0.000000
15 0.000000
16 0.000000
Bonds
1 1 1 2
2 1 1 6
3 2 1 7
4 3 2 3
5 4 2 8
6 4 2 9
7 1 4 3
8 4 3 10
9 4 3 11
10 1 4 5
11 2 4 12
12 3 5 6
13 4 5 13
14 4 5 14
15 4 6 15
16 4 6 16
Angles
1 1 2 1 6
2 2 2 1 7
3 2 6 1 7
4 3 1 2 3
5 4 3 2 8
6 4 3 2 9
7 5 1 2 8
8 5 1 2 9
9 6 8 2 9
10 3 4 3 2
11 4 2 3 10
12 4 2 3 11
13 5 4 3 10
14 5 4 3 11
15 6 10 3 11
16 1 3 4 5
17 2 3 4 12
18 2 5 4 12
19 3 4 5 6
20 5 4 5 13
21 5 4 5 14
22 4 6 5 13
23 4 6 5 14
24 6 13 5 14
25 3 1 6 5
26 4 5 6 15
27 4 5 6 16
28 5 1 6 15
29 5 1 6 16
30 6 15 6 16
Dihedrals
1 1 6 1 2 3
2 2 6 1 2 8
3 2 6 1 2 9
4 3 7 1 2 3
5 4 7 1 2 8
6 4 7 1 2 9
7 1 2 1 6 5
8 2 2 1 6 15
9 2 2 1 6 16
10 3 7 1 6 5
11 4 7 1 6 15
12 4 7 1 6 16
13 5 1 2 3 4
14 6 1 2 3 10
15 6 1 2 3 11
16 6 4 3 2 8
17 7 8 2 3 10
18 7 8 2 3 11
19 6 4 3 2 9
20 7 9 2 3 10
21 7 9 2 3 11
22 1 5 4 3 2
23 3 12 4 3 2
24 2 5 4 3 10
25 4 12 4 3 10
26 2 5 4 3 11
27 4 12 4 3 11
28 1 3 4 5 6
29 2 3 4 5 13
30 2 3 4 5 14
31 3 12 4 5 6
32 4 12 4 5 13
33 4 12 4 5 14
34 5 4 5 6 1
35 6 4 5 6 15
36 6 4 5 6 16
37 6 1 6 5 13
38 7 13 5 6 15
39 7 13 5 6 16
40 6 1 6 5 14
41 7 14 5 6 15
42 7 14 5 6 16
Impropers
1 1 2 1 6 7
2 1 3 4 5 12