Hello everyone.
I have been pondering this for the past few weeks, and I’ve brought something that I’m still unsure about.
Of course, this might be basic content, but… even after looking at other people’s Q&A, there are many parts that are not well understood or are unclear.
1. DOS data of GGA and r2SCAN calculation data
In the case of Density of states, I obtain an energy and density database in the following manner, which is then used for plotting graphs.
76 dos = mpr.get_dos_by_material_id(mid)
77 energies = dos.energies # numpy array
78 densities = dos.get_densities() # numpy array
79 efermi = dos.efermi
80 idx = (abs(energies - efermi)).argmin()
81 dos_at_fermi = densities[idx]
The code for obtaining Entry energy and Cohesive energy was used as follows.
Unlike Cohesive, I understand that Entry values can differ between GGA and r2SCAN.
49 # Get GGA energy
50 thermo_docs = mpr.materials.thermo.search(material_id)
51 for thermo_doc in thermo_docs:
52 if "GGA" in thermo_doc.entries:
53 gga_energy_per_atom = thermo_doc.entries["GGA"].uncorrected_energy_per_atom # GGA energy per atom
54 total_energy = thermo_doc.entries["GGA"].uncorrected_energy # total energy in GGA
55 task_id = next(prop.task_id for prop in thermo_doc.origins if prop.name == "energy")
56 composition = thermo_doc.composition.as_dict()
265 # Get Cohesive energy
266 cohesive_energy = mpr.get_cohesive_energy(
267 material_ids=[material_id], normalization="atom" # normalize with number of atoms
268 )[material_id] # Results are returned as dictionary
In DOS, it is thought that there would be different values depending on the r2SCAN and GGA methods. What calculation method is the DOS in the database based on?
Since Materials Project uses meta-GGA, are you using the calculated values from r2SCAN calculations for GGA structural optimization?
2. Normalization through Volume
I want to normalize the DOS through volume, and to do this, I need to know the structure of the cell used during calculation to add to the database. Are these calculation results from a primitive cell or a conventional cell?
Most data structures that can be obtained from the Materials Project website (to the extent of my knowledge, all of them) have a conventional cell structure. Would it be correct to use the volume of the conventional cell for normalization?
After seeing another user’s query and modifying the code to obtain the following data, I’m unsure what it means. In cases where both Conventional and Primitive are True, is the data uploaded based on a Conventional or Primitive structure?
Are DOS and BAND data based on primary cells or conventional cells?
| Formula (Conventional structure) | MPID | DOS_Primitive | DOS_Conventional | BS_Primitive | BS_Conventional |
|---|---|---|---|---|---|
| Al1Co1 | mp-284 | True | True | True | True |
| Be1Co1 | mp-2773 | True | True | True | True |
| Be20Co4 | mp-1071690 | True | False | True | False |
| Be6Co2 | mp-1183423 | True | False | True | False |
| Ce8Co16 | mp-1112 | True | False | True | False |
| Co12B4 | mp-20373 | True | True | True | True |
| Co12S18 | mp-1183728 | True | False | True | False |
| Co12Se16 | mp-20456 | True | False | True | False |
| Co1I2 | mp-569610 | True | False | True | True |
3. Difference between DOS calculated directly and DOS extracted from database
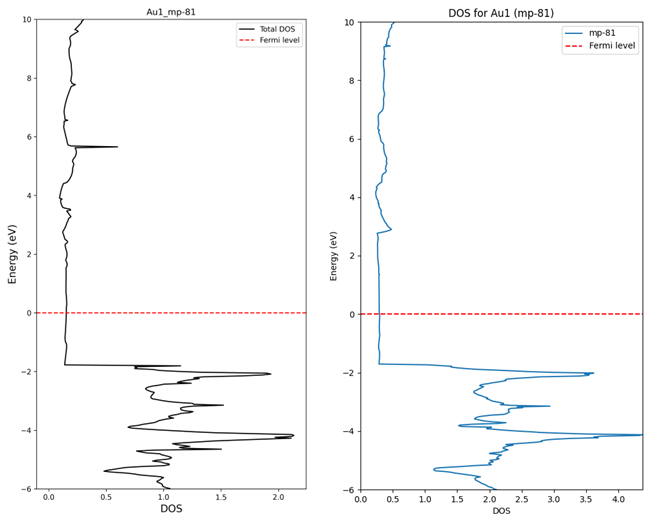
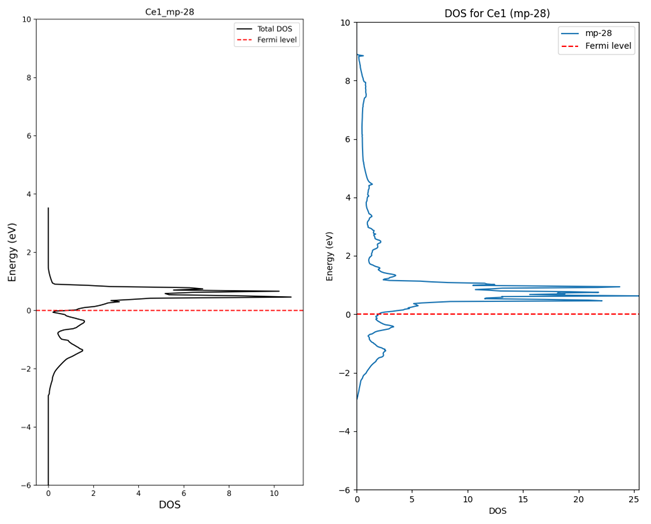
(The data above is raw data from API. Not normalized)
I understand that there can be differences in the graph shape because the values are extracted from SCF calculation results.
(The reason for extracting from SCF is that rough data was needed for pre-screening.)
However, the actual density values appear lower than those obtained through the API. What is the reason for this?
I am grateful to those who diligently answer my often insufficient questions.
I always end up writing long texts, and I am thankful that you take the time to read them with interest.
