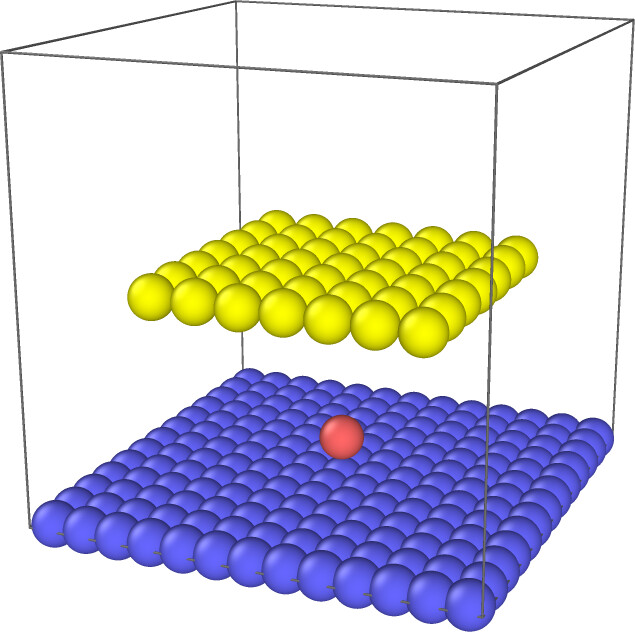
Is there an approach to dynamic update rigid body during the simulation? What I want to do is, in the beginning, I have a base or reaction site(a set of atoms with type 1). At a certain frequency, when a specific type(let’s say type 2) of an atom is close enough to the reaction site, turn the type 2 atom to type 1, and bind those atoms to a new rigid. This process is something like dendrite growing.
Can I use the built-in commands in lammps to do this?
Thanks in advance that helping me with this freakish question
Well, first a question. It seems that you may be mixing up “rigid” with “immobile”. Do you want to have all type 1 atoms moving as one body, or do you want those atoms not move at all?
Then you should not use the term “rigid”. A rigid body can move but all atoms retain their relative positions. Immobile atoms do not move.
Now, about your problem. Actually, you have two problems: 1) how to detect when an atom should be switched from type 2 to type 1, and 2) how to change which atoms are mobile and which are immobile.
For problem 1) you could use fix bond/create. It will add a bond as a side effect, but that can be ignored here. But it also adds the option to change the type of an atom if the bond is added.
For problem 2) you can use time integration (e.g. with fix nve) on a dynamic group that only contains type 2 atoms. If there is no time integration for type 1 atoms, they are immobile.
I am so glad that fix bond/create meet my request. But I also have two questions:
What does the meaning of extra/special/per/atom. I know the command special_bond fene, but I don’t know how to determine the number of extra/special/per/atom here.
When the atom changes its type, it seems that the group does not update. I think it needs to use a dynamic group. According to the manual, dynamic group can not simply use type keyword, but region, variable and property. I doesn’t figure out how to use these three keywords to specify the atom type.
I am so glad that you can help me with it. It almost works!
Here is my script that can reproduce what I want to do
units lj
atom_style full
atom_modify map array
region box block -5 5 -5 5 0 10
boundary p p f
create_box 3 box bond/types 1 extra/bond/per/atom 5 extra/special/per/atom 50
# --- reaction site ---
create_atoms 1 single 0 0 1
mass 1 1.0
# --- setup wall --- type 1
region wall block INF INF INF INF 0.0 0.5
lattice sc 2.0
create_atoms 2 region wall
mass 2 1.0
region antiwall block -3 3 -3 3 5.5 6.0
create_atoms 3 region antiwall
mass 3 1.0
fix zhiwalls all wall/reflect zhi EDGE
# --- potential setup ---
pair_style lj/cut 2.5
pair_coeff 1 3 1.5 1.0 2.5 # particle & reaction site
pair_coeff * * 1.0 1.0 1.0
pair_modify shift yes
bond_style fene
special_bonds fene
bond_coeff * 30.0 1.5 1.0 1.0
timestep 0.002
minimize 1.0e-5 1.0e-6 100000 1000000
group reactionSite type 1 dynamic all every 1 # wrong!
group surface type 2
group particle type 3
velocity particle create 1.0 74982687
fix 1 particle nve
fix 2 particle langevin 1.0 1.0 1000 12345
fix 3 all bond/create 1 1 3 1.0 1 jparam 1 1 # turn
dump obs all custom 10 big.dump id type q x y z
run 10000
In order to apply the special_bond properties, the neighbor list code needs to know which atoms are so-called “special neighbors”, i.e. pairs of atoms that are connected to each other via the bond topology either directly (1-2 special neighbors) or with one or two atoms in between (1-3 and 1-4 special neighbors). For efficiency reasons, the identity of these neighbors needs to be stored with each atom and the space for that needs to be reserved when the simulation box is created. By default, LAMMPS will run a check when the box is created and determine the minimal required number (to save memory). But if you add bonds later, you may need to have more space because the list could be larger for some atoms, and this space is reserved with extra/special/per/atom.
BTW: it doesn’t hurt much to just use a rather large number initially, say 100. Once you have everything working, you can check if you can make it smaller. Whenever LAMMPS updates the special bond information, there is usually some output telling you what the current requirements are. So it is easy to adjust later. In most cases, the extra memory doesn’t really hurt much. The amount of available RAM per processor these days is much larger than 20+ years ago when the core of LAMMPS was conceived and saving memory was paramount when you wanted to run parallel calculations of very large systems with very many atoms.
Correct.
You need to think a bit indirectly. You need a yes/no information for each atom (i.e. zero and non-zero) and you have access to almost every property through atom style variables. Rather than having to add support for lots of different settings and properties (which is a lot of work and requires maintenance), we added support for variables. Something like this:
variable istype1 atom type==1
variable istype2 atom type==2
My supervisor asks me the last question, say, if there are positive and negative ions in the system, if one ion is gradually converted to another atom type, is there any way to make the counterion disappear (delete) synchronously?
That would require using fix adapt and you would have to define a group that fix adapt is applied to. Supported coulomb pair styles export a per-atom “scale” property and thus you could scale the charge for the atoms in the selected groups. I am quoting this from memory, so I advise to carefully study the corresponding documentation. If you need the compute the free energy associated with that process, things get a bit more complex and you need to vanish the charge and the LJ properties separately. Details of how to do this are far beyond the scope of this forum. There is some Howto information in the LAMMPS manual, some examples and references to related publications. If you search through the archives you may also find some suggestion for text books related to that.
I think adapting to scale the charge is hard to implement.
What about simply deleting the atoms? For example, I compute the number of atomtype 1 turn into another type and delete its counterion by using this number. I don’t remember if there is a command that can dynamically delete atoms with a changed number.
Scaling charges is required with ions. Otherwise the local change would be like the effect of a microscopic atomic bomb.
I am not aware of a facility in LAMMPS to detect and select a corresponding counterion and eliminate it in a synchronized fashion. This would require writing a new fix that combines features from fix bond/create, fix adapt, and fix gcmc, if my ad hoc thinking is correct.
I understand. Thanks for your help. If I need to do so in the future I may write a new fix command. Those things quite satisfy me. Lammps is the best and most flexible MD package ever.