Hi, Alex
I am a beginner in amset. I have noticed that there exist methods for calculating elastic constants, dielectric constants, deformation potential for materials. however, unfortunately, I have not found a signal to help me calculate the electric conductivity. Can you give me some guidance? Your help would be highly appreciated.
Best wishes,
Leo
Hello,
The electronic conductivity is determined automatically in AMSET by using the dielectric, deformation, and other properties as inputs. If you follow the guidance in the documentation it should output the electronic conductivity along with the carrier mobility.
Hi,Steven,
I am glad to hear that the electric conductivity can be obtained, but can you tell me where the files, to say, the electronic conductivity and the carrier mobility are? I have not found this in my OUTCAR file.
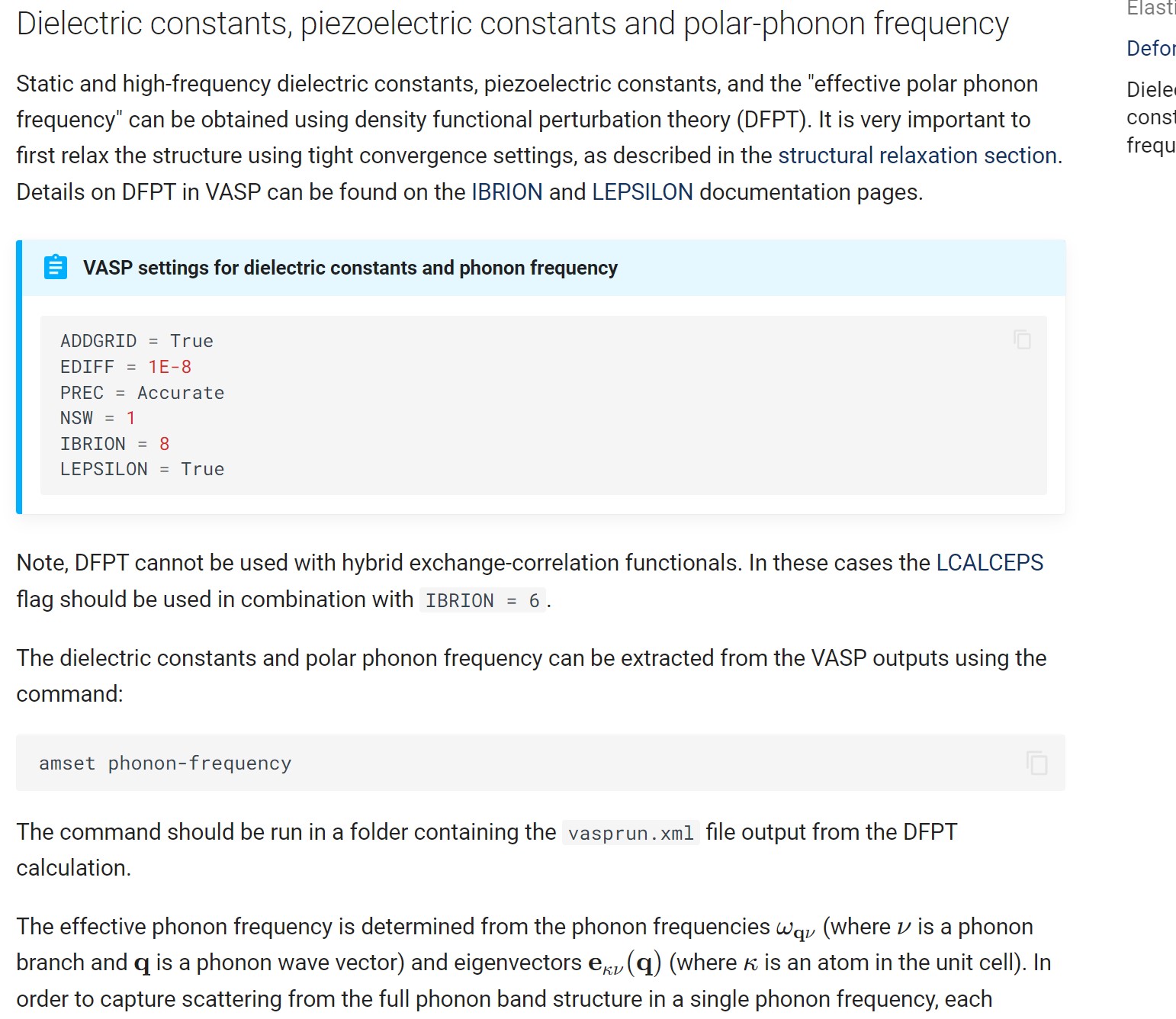
Your help would be highly appreciated.
Hi Leo
You won’t find that in your OUTCAR file. Follow the steps in the amset documentation.
In the last step, when you run “amset run” then only you will get all the required information.
Regards
Zeeshan
Yes, the conductivity should be in the amset.log file produced by AMSET.
Hi, Steven
Long time no see, I recently began to analyze the properties of Si. But when I click the codes" amset run" into my PC, it seems that something was wrong with my process. I have not found out what caused this problem, could you help me? Your help would be highly appreciated!
Hi. @LeoLiu, Please can you paste the full output of the amset log file.


Hi, Alex
Chances are that I made some mistakes in my calculation, but I could not find out them. Thank you for helping me solve this problem. It reads “new users can only post 3 embedded media items one time.” So I just divide the full one into two parts. Looking forward to your reply soon!
Best wishes!



Hi @LeoLiu
It is very hard to tell what is going on in the log file from the screenshots. Please can you copy and paste the text directly (not an image)?
Best,
Alex
Hi,Alex.
I just forgot I can paste the log.file. Sorry for the unnecessary trouble. The following are the codes. Best wishes!
/$$$$$$ /$$ /$$ /$$$$$$ /$$$$$$$$ /$$$$$$$$
/$$__ $$| $$$ /$$$ /$$__ $$| $$_____/|__ $$__/
| $$ \ $$| $$$$ /$$$$| $$ \__/| $$ | $$
| $$$$$$$$| $$ $$/$$ $$| $$$$$$ | $$$$$ | $$
| $$__ $$| $$ $$$| $$ \____ $$| $$__/ | $$
| $$ | $$| $$\ $ | $$ /$$ \ $$| $$ | $$
| $$ | $$| $$ \/ | $$| $$$$$$/| $$$$$$$$ | $$
|__/ |__/|__/ |__/ \______/ |________/ |__/
v0.4.11
A. Ganose, J. Park, A. Faghaninia, R. Woods-Robinson,
A. Jain, in prep.
amset starting on 08 Mar 2022 at 22:20
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ SETTINGS ~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Run parameters:
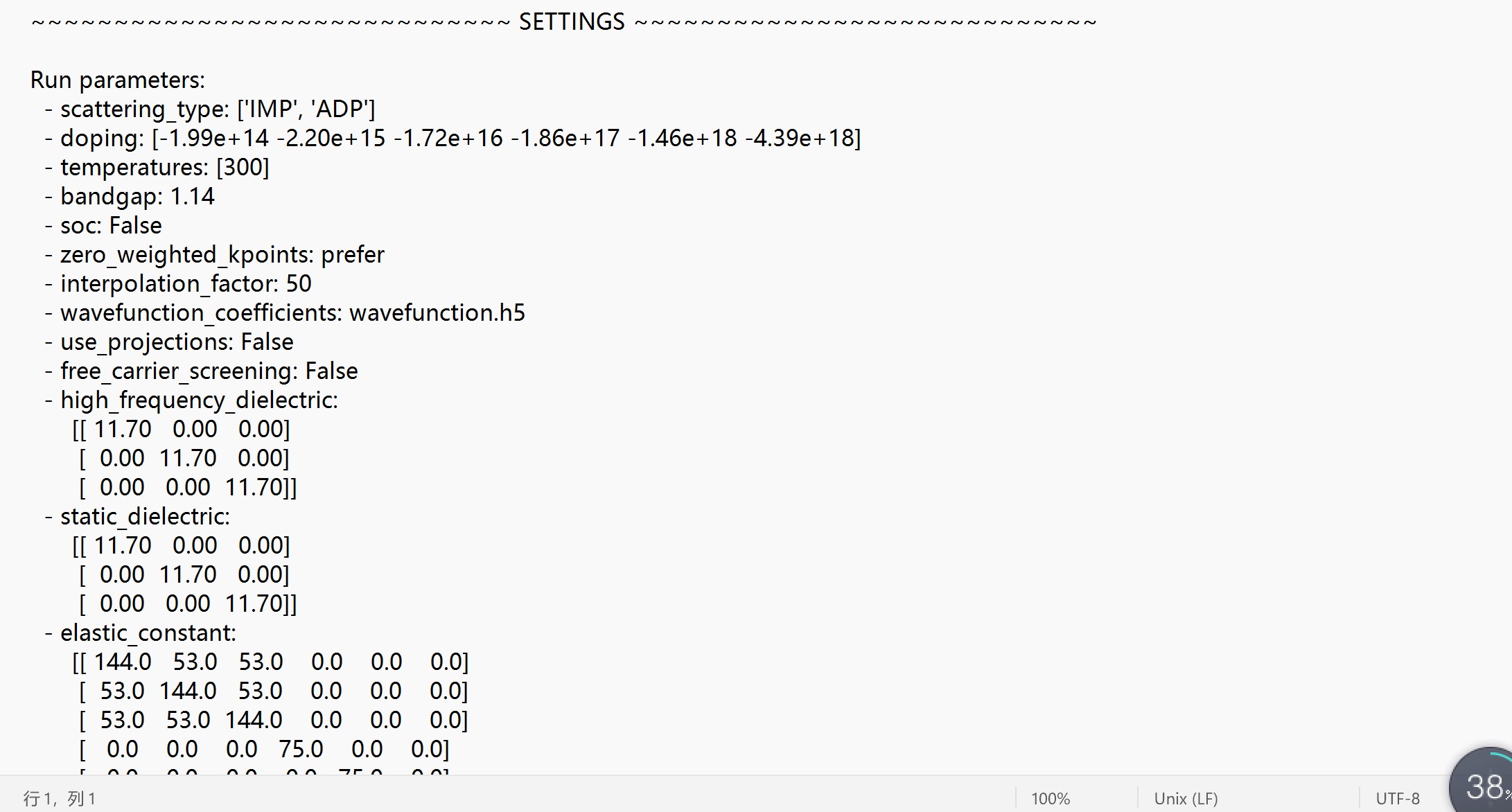
- scattering_type: ['IMP', 'ADP']
- doping: [-1.99e+14 -2.20e+15 -1.72e+16 -1.86e+17 -1.46e+18 -4.39e+18]
- temperatures: [300]
- bandgap: 1.14
- soc: False
- zero_weighted_kpoints: prefer
- interpolation_factor: 50
- wavefunction_coefficients: wavefunction.h5
- use_projections: False
- free_carrier_screening: False
- high_frequency_dielectric:
[[ 11.70 0.00 0.00]
[ 0.00 11.70 0.00]
[ 0.00 0.00 11.70]]
- static_dielectric:
[[ 11.70 0.00 0.00]
[ 0.00 11.70 0.00]
[ 0.00 0.00 11.70]]
- elastic_constant:
[[ 144.0 53.0 53.0 0.0 0.0 0.0]
[ 53.0 144.0 53.0 0.0 0.0 0.0]
[ 53.0 53.0 144.0 0.0 0.0 0.0]
[ 0.0 0.0 0.0 75.0 0.0 0.0]
[ 0.0 0.0 0.0 0.0 75.0 0.0]
[ 0.0 0.0 0.0 0.0 0.0 75.0]]
- deformation_potential: deformation.h5
- defect_charge: 1
- compensation_factor: 2
- energy_cutoff: 1.5
- fd_tol: 0.05
- dos_estep: 0.01
- symprec: 0.01
- nworkers: -1
- cache_wavefunction: True
- calculate_mobility: True
- separate_mobility: True
- mobility_rates_only: False
- file_format: json
- write_input: False
- write_mesh: True
- print_log: True
- write_log: True
~~~~~~~~~~~~~~~~~~~~~~~~~~~~ STRUCTURE ~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Structure information:
- formula: Si
- # sites: 2
- space group: Fd3-m
Lattice:
- a, b, c [angstrom]: 5.77, 5.77, 5.77
- a, b, y [deg]: 60, 60, 60
~~~~~~~~~~~~~~~~~~~~~~~~~~ BAND STRUCTURE ~~~~~~~~~~~~~~~~~~~~~~~~~
Input band structure information:
- # bands: 10
- # k-points: 1
- Fermi level: -0.888 eV
- spin polarized: False
- metallic: False
Band gap:
- direct band gap: 1.413 eV
- direct k-point: [0.00, 0.00, 0.00]
Valence band maximum:
- energy: -0.903 eV
- k-point: [0.00, 0.00, 0.00]
- band indices: 3, 4, 5
Conduction band minimum:
- energy: 0.510 eV
- k-point: [0.00, 0.00, 0.00]
- band indices: 6, 7, 8
~~~~~~~~~~~~~~~~~~~~~~~~~~ INTERPOLATION ~~~~~~~~~~~~~~~~~~~~~~~~~~
Getting band interpolation coefficients
- time: 0.0288 s
Interpolation parameters:
- k-point mesh: 19x19x19
- energy cutoff: 1.5 eV
Interpolating spin-up bands 3-8
- time: 0.0749 s
bandgap set to 1.140 eV, applying scissor of -0.273 eV
Generating tetrahedron mesh vertices
- time: 0.0679 s
Initializing tetrahedron band structure
- time: 0.0292 s
Initializing momentum relaxation time factor calculator
Initializing wavefunction overlap calculator
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ DOS ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
DOS parameters:
- emin: -0.77 eV
- emax: 0.37 eV
- dos weight: 2
- n points: 114
Generating tetrahedral DOS:
- time: 0.0070 s
Intrinsic DOS Fermi level: -0.1965 eV
DOS contains 0.000 electrons
Calculated Fermi levels:
conc [cm-3] temp [K] E_fermi [eV]
------------- ---------- --------------
-1.99e+14 300.0 -151.3708
-2.20e+15 300.0 -151.3708
-1.72e+16 300.0 -151.3708
-1.86e+17 300.0 -151.3708
-1.46e+18 300.0 -151.3708
-4.39e+18 300.0 -151.3708
ERROR: amset exiting on 08 Mar 2022 at 22:20
Traceback (most recent call last):
File "/opt/ohpc/pub/anaconda3/envs/amset/bin/amset", line 10, in
<module>
sys.exit(cli())
File "/opt/ohpc/pub/anaconda3/envs/amset/lib/python3.9/site-
packages/click/core.py", line 829, in __call__
return self.main(*args, **kwargs)
File "/opt/ohpc/pub/anaconda3/envs/amset/lib/python3.9/site-
packages/click/core.py", line 782, in main
rv = self.invoke(ctx)
File "/opt/ohpc/pub/anaconda3/envs/amset/lib/python3.9/site-
packages/click/core.py", line 1259, in invoke
return _process_result(sub_ctx.command.invoke(sub_ctx))
File "/opt/ohpc/pub/anaconda3/envs/amset/lib/python3.9/site-
packages/click/core.py", line 1066, in invoke
return ctx.invoke(self.callback, **ctx.params)
File "/opt/ohpc/pub/anaconda3/envs/amset/lib/python3.9/site-
packages/click/core.py", line 610, in invoke
return callback(*args, **kwargs)
File "/opt/ohpc/pub/anaconda3/envs/amset/lib/python3.9/site-
packages/amset/tools/run.py", line 139, in run
runner.run()
File "/opt/ohpc/pub/anaconda3/envs/amset/lib/python3.9/site-
packages/amset/core/run.py", line 62, in run
mem_usage, (amset_data, usage_stats) = memory_usage(
File "/opt/ohpc/pub/anaconda3/envs/amset/lib/python3.9/site-
packages/memory_profiler.py", line 336, in memory_usage
returned = f(*args, **kw)
File "/opt/ohpc/pub/anaconda3/envs/amset/lib/python3.9/site-
packages/amset/core/run.py", line 114, in _run_wrapper
amset_data, dos_time = self._do_dos(amset_data)
File "/opt/ohpc/pub/anaconda3/envs/amset/lib/python3.9/site-
packages/amset/core/run.py", line 247, in _do_dos
amset_data.calculate_fd_cutoffs(
File "/opt/ohpc/pub/anaconda3/envs/amset/lib/python3.9/site-
packages/amset/core/data.py", line 289, in calculate_fd_cutoffs
cmin, cmax = get_min_max_cutoff(weight_cumsum)
File "/opt/ohpc/pub/anaconda3/envs/amset/lib/python3.9/site-
packages/amset/core/data.py", line 272, in get_min_max_cutoff
min_idx = np.where(cumsum < fd_tolerance / 2)[0].max()
File "/opt/ohpc/pub/anaconda3/envs/amset/lib/python3.9/site-
packages/numpy/core/_methods.py", line 39, in _amax
return umr_maximum(a, axis, None, out, keepdims, initial,
where)
ValueError: zero-size array to reduction operation maximum which
has no identity
Hi @LeoLiu,
I can see your input electronic structure only has a single k-point. For the silicon primitive cell with only 2 atoms that is far too small. You will typically need 10x10x10 or more k-points in order to get a reasonable interpolation for small unit cells.
I think if you increase the k-point grid for the DFT calculation that will fix your problem.
Best,
Alex
Hi, Alex
Sorry for troubling you so much as a freshman in using amset. I made some adjustments as you told me, namely, I changed the k-points from 111 to 111111, but when I did some instructions, like “amset wave” it read the first codes,
ValueError: Incorrect value of vasp_type given (None). Please open an issue if you are certain this WAVECAR was generated with the given vasp_type.
and when it comes to the calculations of deformation potential, it read the second ones,
ValueError: Expected 1331 points but found 469
I was wondering whether I made some mistakes. And the screen shot is my kpoints file.

Looking forward to your reply!
Best wishes!
Hmm that is unusual.
I noticed something odd about your structure. You are running a silicon cell with 2 atoms. The lattice constant is 5.77 Å which is the right lattice constant for the conventional cell, but your cell angles are 60 degrees which (along with the number of atoms in the cell) indicates a primitive cell. I would double check that you are using the right structure. You can download it from the Materials Project to check.
If that doesn’t help, please can you try a slightly smaller k-point mesh as an additional test, for example say 8x8x8?
Hi, Alex
I have downloaded the conventional cell POSCAR from the MP, and made the value of kpoints smaller, 888, but I still encountered a case never seen before, I was wondering if I made a mistake or there is some bug in it. Looking forward to your reply.
Here is what amset reads when I instructed" amset wave." ValueError: Incorrect value of vasp_type given (None). Please open an issue if you are certain this WAVECAR was generated with the given vasp_type.
Best wishes!
Please could you send me your OUTCAR, vasprun.xml, WAVECAR, and POTCAR files and I will take a look.
I found out that this is a bug that was fixed in the latest version of pymatgen. If you update pymatgen and try amset wave again it should work!
Hi, Alex
Just as you said, I finally solved this problem. Thank you so so so so so so much for helping me.
Best wishes!
Hi, Alex
Thanks for helping me resolve the problem last time. Recently I found that the relaxation time, even the average one, did not be presented in my amset.log file. I did not know if I made some mistakes. Additionally, I was wondering if you could add some codes to help calculate the electronic thermal conductivity rate, that is nice of you!
Best regards.