Dear Kasja,
The best way to look at the polyethylene.emc structure is to consider it as a sort of function, which can be used in conjunction with preset variables – either of which can be provided on the command line or as part of a wrap-around input script (also typically with a .emc extension). EMC operates in two modes:
- Script mode in which the first line of
.emc reads (* EMC: Script *),
- Structural input/output mode in which the first line of
.emc reads (* EMC: Enhanced Monte Carlo simulations *); this format is normally used as restart exchange format and gzipped by default.
The easiest way to see the difference between the two modes is to go through the exercise of a simple EMC Setup example (create setup.esh with emc_template.sh chemistry followed by execution of setup.esh). This EMC Setup .esh script generates build.emc by default, which can either be executed manually by emc.sh build.emc, or automatically by adding emc_execute true to the ITEM OPTIONS section.
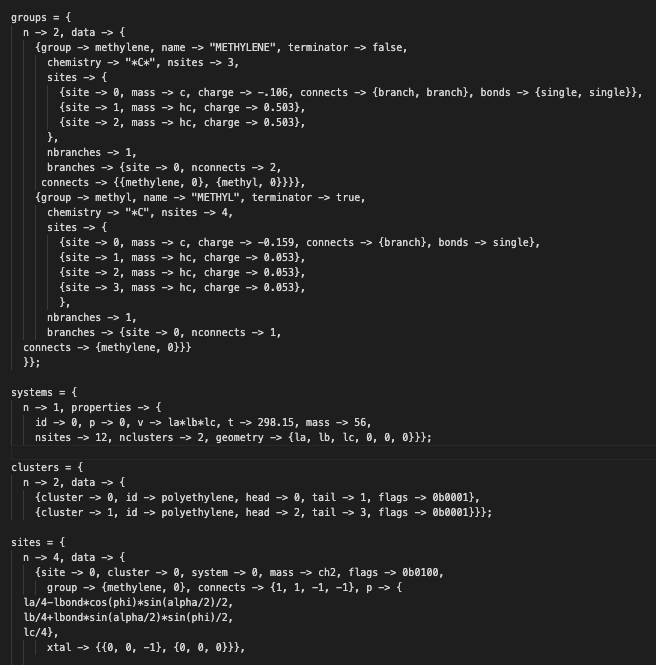
A group – part of the (* Groups *) section – represents a repeat unit, which defines the chemical environment of a site. This chemical environment in groups is used to determine types during the force field typing process. These resulting types are then transferred to each site in the (* Sites *) section. The (* Clusters *) section maintains the beginning and end site of a cluster (which equals a molecule in atomistic representations). The connects keyword of a site in the (* Sites *) section defines the connectivity of all sites in a cluster. Internally, recursive algorithms are used to determine all member sites of a cluster. The choice for determining member sites recursively stems from Monte Carlo simulations being able to make and break bonds during the simulation. Maintaining continuously altering lists of member sites is thought of being inefficient.
Having said all this, I would suggest using an InsightII unit cell for crystalline polyethylene, which contains connectivity and use this to import a structure using EMC Setup, since direct alteration of EMC Script files tends to be tedious.
A simple EMC Setup .esh could look like
# Options section
ITEM OPTIONS
replace true
field pcff
mass true
ntotal 2000
density 0.85
expert true
emc_execute true
ITEM END # OPTIONS
# Variables section
ITEM VARIABLES spot=0
nx 4
ny 5
nz 10
nrepeat 10
ITEM END # VARIABLES
# Groups section
ITEM GROUPS
ethylene *CC*,1,ethylene:2
methyl *C,1,ethylene:1, 1,ethylene:2
ITEM END # GROUPS
# Clusters section
ITEM CLUSTERS
surface import, name="ethylene.car", &
type=surface, percolate=true, formal=false, &
ncells=nx:ny:nz, field=pcff
polymer alternate,1
ITEM END # CLUSTERS
# Polymers section
ITEM POLYMERS
polymer
1 ethylene,nrepeat,methyl,2
ITEM END # POLYMERS
for which you need the following two input structures
ethylene.car (1.1 KB), and
ethylene.mdf (1.4 KB),
to be installed in the same directory as
setup.esh (830 Bytes).
This creates a crystal with polyethylene molecules parallel to the surface, above which oligomeric amorphous ethylene consisting of 22 carbons is grown. As an aside: please use a fresh version of EMC (uploaded at or after April 16, 2025) since I noticed a mistake in importing InsightII structures.
Best,
Pieter