I assume you have not defined the density. By default, the density is set to 1 g/cc. You can lower this density to say 0.1 g/cc if you want to build one structure in a much larger box. In EMC Setup you would do this by setting density under ITEM OPTIONS to 0.1. I am not quite sure how to do this when using PyEMC. Just out of curiosity: are you planning on calculating Tg based on a single polymer/oligomer molecule, or are you wanting to deduce Tg from a density vs. temperature plot of polymers at their condensed phase density? Just for your information: EMC includes a Tg determination example under examples/setup/environment/t_glass.
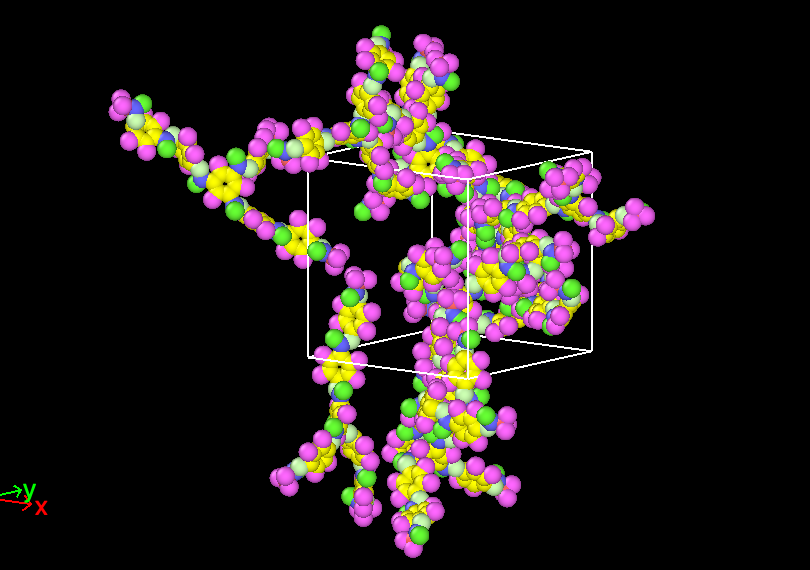
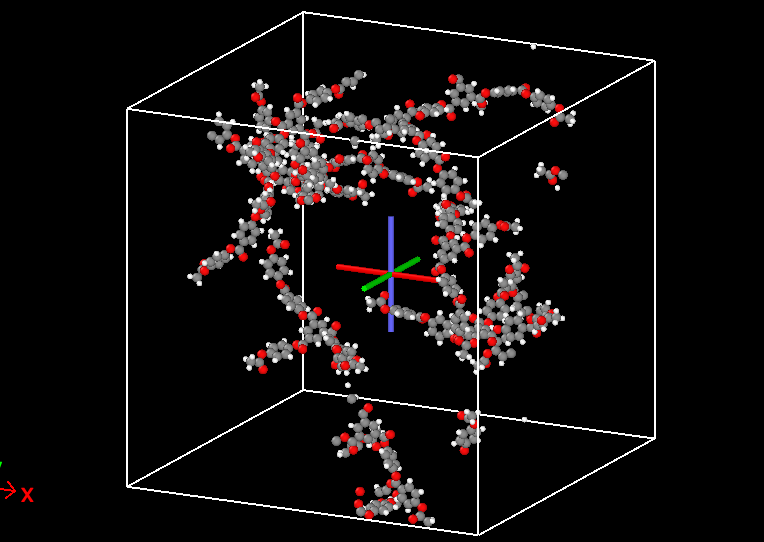
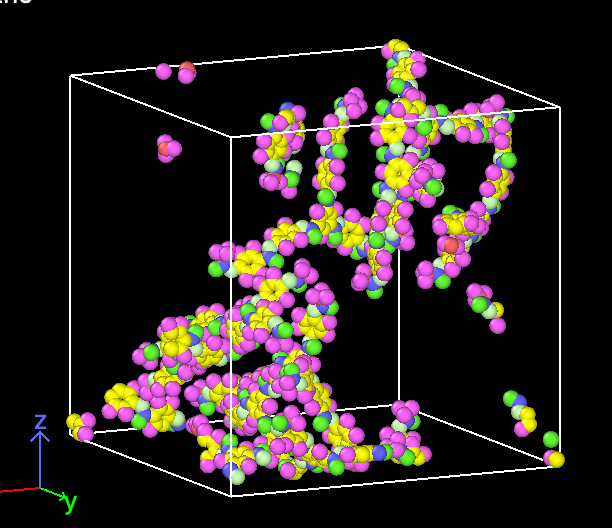
I have played around with the density setting, and it still seems to still build outside the box even when I choose something like density = 0.01.
I thought it might have something to do with the ‘system_pbc’ setting, but this also did not change anything… Hmm.
I was able to manually rewrite the data file with a little python script to produce what I thought system_pbc would do:
My plan was to compute Tg from a temperature/density plot after annealing a sample of several oligomers. I was not aware there was another way.
I will take a look at that example, that sounds mighty useful! I saw it there before but I haven’t figured out how to get it to work yet… I had issues with the ${dir} variable (it makes a folder called …/a-pp/200 205 210…300 instead of separate ones and then the script thinks they’re separate arguments) and also the atom.sh script isn’t able to find the .pl scripts even though I think I added the ‘scripts’ and ‘bin’ folders to my PATH. Is this something wrong with my setup file, or an issue with my installation?
Well… Boxes always assume periodic images. This means that whatever looks to be outside of your box will actually be inside the box, since it is mapped back in when the periodic image convention is applied. I assume you are using .pdb and .psf for your visualization. Set the options pdb_unwrap and pdb_pbc under ITEM OPTIONS to false if you do not want to unwrap your polymer and you want to apply periodic boundary conditions to the box you visualize.
Having said that, you will want to simulate at condensed matter conditions. This means that your density should be around 1 g/cc and not at very low conditions like 0.01 g/cc.
I am not sure which ${dir} variable in what file you are referring to. Could you be more specific? Also, could you tell me what operating system you are using?
The ${dir} variable I am referring to is in this line 325 of ./run/2023072600.sh:
run_emc() {
local dir=“$1”; shift;
printf “### ${dir}\n\n”;
if [ ! -e ${dir} ]; then
run mkdir -p ${dir};
fi; `
It looks like it is supposed to be set by this line in the same file: dir="data/${STAGE}/${TRIAL}/${TEMPERATURE}/${COPY}";
and I’m pretty sure that’s what’s causing that very first error. The further errors (“invalid preprocessing directive” and “can’t find emc_”) are completely mysterious to me, but a lot of them seem vaguely directory-related
Thank you for the logs. Did you use emc.pl or emc_setup.pl to generate ./run/2023072600.sh? Do you have access to a queueing system on the machine you started ./run/2023072600.sh from?
I believe I have fixed the strange filesystem creation problem!! However I couldn’t tell you what exactly was wrong. After clearing everything out of my PATH variable and reinstalling EMC it just worked suddenly.
I used emc_setup.pl to generate that last run script.
emc.pl generates some different errors but actually fewer! 2023072600_emc_setup.log (166.9 KB) 2023072600_emc.log (79.4 KB)
I do have access to an HPC with slurm, but I have been trying to get as much building and troubleshooting done locally as possible.