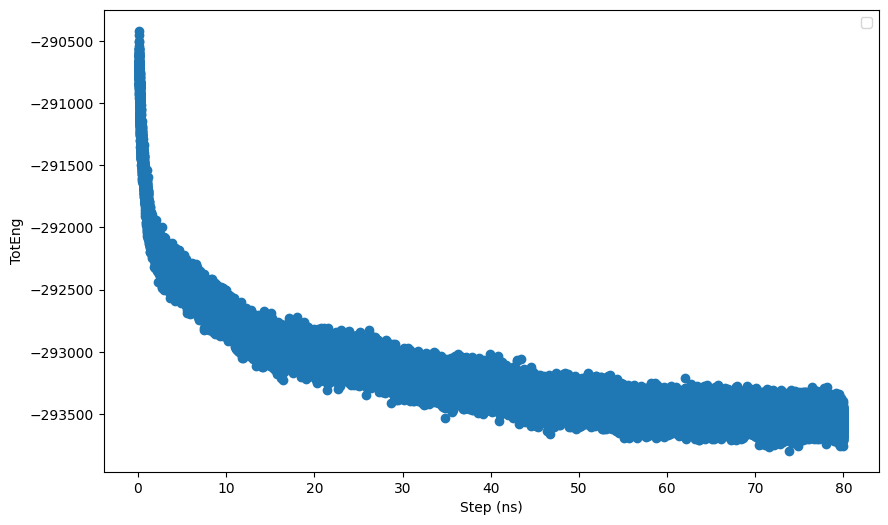
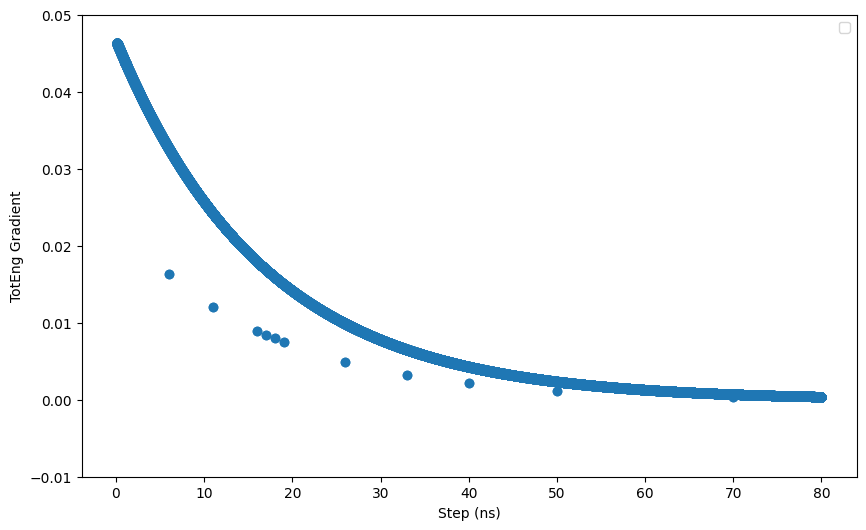
Hi all,
I am running a npt simulation of a system consisting a nanoparticle (NP) immeresed in a solvent mixture (contains salts and organic solvents). I noticed that when I continue the simulation using restart files from the previous run, the potential energy and columbic pairwise energy do not start from the same value from the previous run. I have attached a short input script and the log files. I also noticed that when I exclude the interactions between the NP using the neigh_modify command, the differnces between the energies from the restart file and the previous run is larger. I believe the problem lies in the kspace solver but not sure what it is exactly. Hope someone can help. Thanks.
Vaishali
include "../system.in.init"
read_data "../system.data"
# read_restart test.restart.500
include "../system.in.settings"
group NP type 1 2 3 4 5 6
group LP30 type 7 8 9 10 11 12 13 14 15 16 17 18 19 20
group OH type 1 4
neigh_modify exclude group NP NP
print "--------- beginning simulation (using fix npt) ---------"
velocity LP30 create 300 49245 rot yes mom yes dist gaussian
dump 1 all custom 1000 traj_npt2.lammpstrj id mol type x y z ix iy iz
thermo_style custom step pe etotal evdwl ecoul temp press vol
thermo 500
restart 500 test.restart
fix fxMobile LP30 npt temp 300 300 100 iso 1.0 1.0 1000 drag 1 dilate LP30
compute tempMobile LP30 temp
fix_modify fxMobile temp tempMobile
timestep 1.0
run 500
the log file:
PPPM initialization ...
using 12-bit tables for long-range coulomb (src/kspace.cpp:342)
G vector (1/distance) = 0.27818548
grid = 90 90 90
stencil order = 5
estimated absolute RMS force accuracy = 0.00034430813
estimated relative force accuracy = 1.0368737e-06
using double precision FFTW3
3d grid and FFT values/proc = 140608 97200
Generated 190 of 190 mixed pair_coeff terms from arithmetic mixing rule
WARNING: Neighbor exclusions used with KSpace solver may give inconsistent Coulombic energies (src/neighbor.cpp:654)
Neighbor list info ...
update: every = 1 steps, delay = 0 steps, check = yes
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 14
ghost atom cutoff = 14
binsize = 7, bins = 12 12 12
1 neighbor lists, perpetual/occasional/extra = 1 0 0
(1) pair lj/cut/coul/long, perpetual
attributes: half, newton on
pair build: half/bin/newton
stencil: half/bin/3d
bin: standard
Per MPI rank memory allocation (min/avg/max) = 58.98 | 59.77 | 61.3 Mbytes
Step PotEng TotEng E_vdwl E_coul Temp Press Volume
0 -124691.18 -85059.223 195945.96 316626.21 280.73691 35184.126 555412.25
500 -299504.45 -186230.92 5556.2468 286587.09 802.38428 -53097.14 449896.5
with restart file generated from the above run:
include "../system.in.init"
# read_data "../system.data"
read_restart test.restart.500
include "../system.in.settings"
group NP type 1 2 3 4 5 6
group LP30 type 7 8 9 10 11 12 13 14 15 16 17 18 19 20
group OH type 1 4
neigh_modify exclude group NP NP
print "--------- beginning simulation (using fix npt) ---------"
# velocity LP30 create 300 49245 rot yes mom yes dist gaussian
dump 1 all custom 1000 traj_npt2.lammpstrj id mol type x y z ix iy iz
thermo_style custom step pe etotal evdwl ecoul temp press vol
thermo 500
restart 500 test.restart
fix fxMobile LP30 npt temp 300 300 100 iso 1.0 1.0 1000 drag 1 dilate LP30
compute tempMobile LP30 temp
fix_modify fxMobile temp tempMobile
timestep 1.0
run 0
the log file from run using restart file:
PPPM initialization ...
using 12-bit tables for long-range coulomb (src/kspace.cpp:342)
G vector (1/distance) = 0.28200344
grid = 90 90 90
stencil order = 5
estimated absolute RMS force accuracy = 0.00028092041
estimated relative force accuracy = 8.4598347e-07
using double precision FFTW3
3d grid and FFT values/proc = 140608 97200
Generated 190 of 190 mixed pair_coeff terms from arithmetic mixing rule
All restart file global fix info was re-assigned
WARNING: Neighbor exclusions used with KSpace solver may give inconsistent Coulombic energies (src/neighbor.cpp:654)
Neighbor list info ...
update: every = 1 steps, delay = 0 steps, check = yes
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 14
ghost atom cutoff = 14
binsize = 7, bins = 11 11 11
1 neighbor lists, perpetual/occasional/extra = 1 0 0
(1) pair lj/cut/coul/long, perpetual
attributes: half, newton on
pair build: half/bin/newton
stencil: half/bin/3d
bin: standard
Per MPI rank memory allocation (min/avg/max) = 61.87 | 62.35 | 63.2 Mbytes
Step PotEng TotEng E_vdwl E_coul Temp Press Volume
500 -304443.06 -191169.54 5556.2468 291640.05 802.38428 -53100.458 449896.5