Hello everyone,
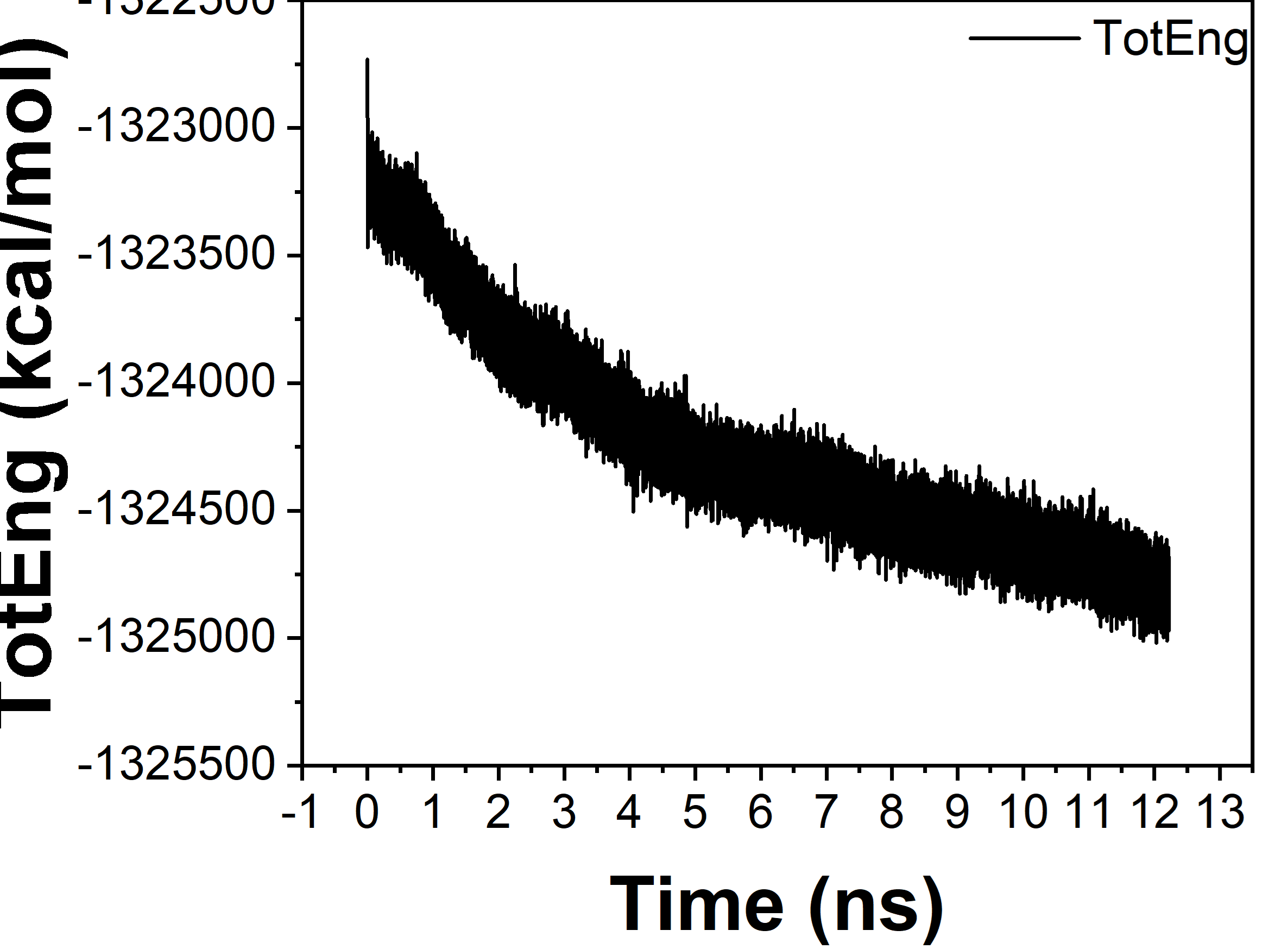
I hope you are doing well. I’m trying to perform a relaxation using a classical forcefield (ClayFF). My unit cell (around 800 atoms) was initially relaxed after 3 ns. However, when I created a supercell (2x2x3), which contains around 10 000 atoms, it is taking almost 13 ns, and the energy still hasn’t stabilized (see figure 1).
I’m wondering if this is just fluctuations, as the changes are not that significant compared to the number of atoms. By recovering a structure at 8 ns, it gives the desired structural and mechanical properties. Do you think this slight decrease could be ignored, or should I wait longer?
figure 1 :
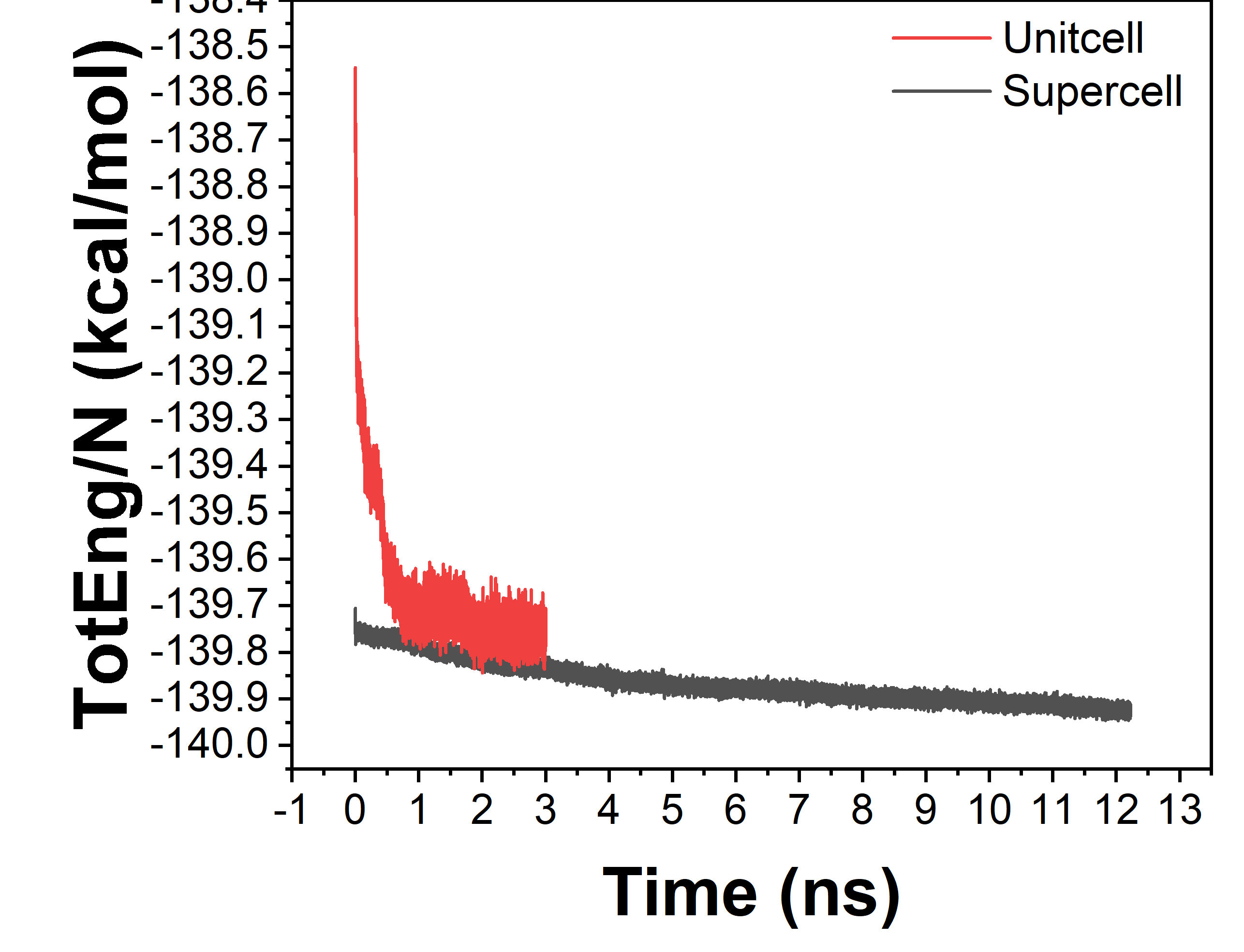
figure 2 :
Thank you very much for your help,
This is impossible to answer in such generality and not knowing anything about the geometry and details of how equilbration is attempted. Also a visualization of the geometries at the beginning and during the equilibration can often provide helpful clues. Finally, it is also possible that your system is “leaking” energy somehow.
Some observations:
- A larger system can have larger fluctuations and thus can explore configurations that are not accessible to a smaller system.
- A decline in total energy is still visible after 3ns in your “unitcell” system. It would be more evident, if you’d use fix ave/time to do a sliding window averaging. So I would not say the system is fully equilibrated at that stage
- you can save time by first running the unit cell system longer, then do a replicate 1 1 3 and continue equilibration, then replicate 1 2 1 and continue and then replicate 2 1 1 and further equilibrate.
What is a good time to stop equilibration is really hard to say since one can only say for certain that a system is not equilibrated. Also, the answer to the question of what is a properly equilibrated system has nothing to do with LAMMPS, but is a question on general MD and thus off-topic for this forum.