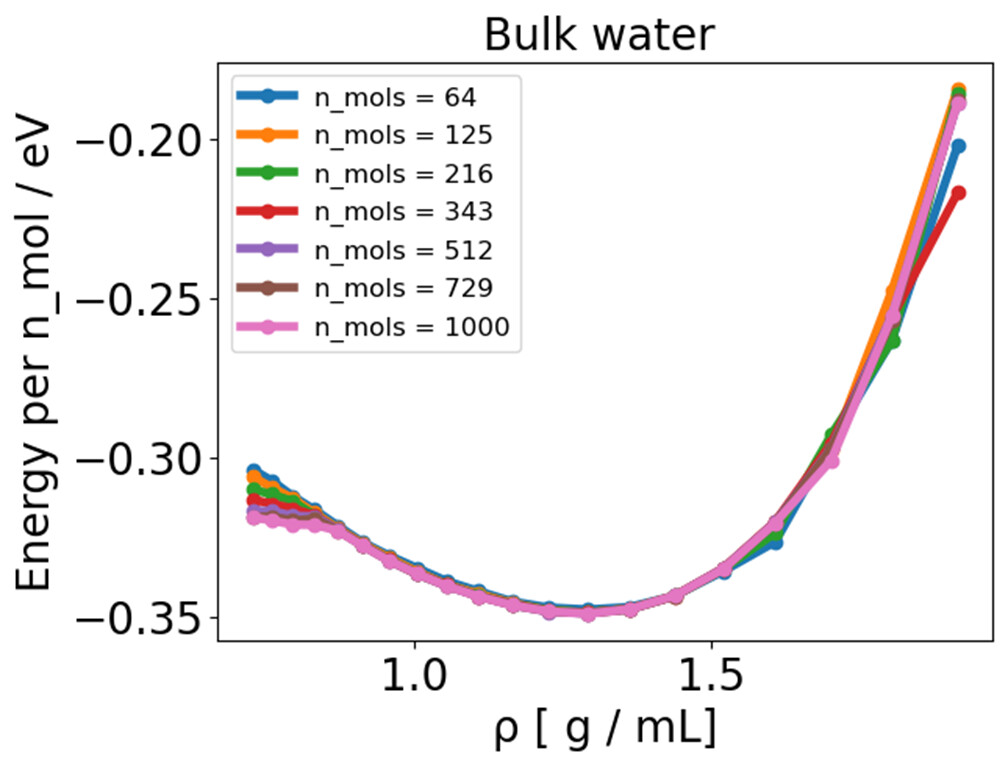
I have been running bulk NVT TIP3P simulations of 64 water molecules where I vary the cell size and keep the number of water molecules constant, giving me different densities and an energy-volume or energy-density curve.
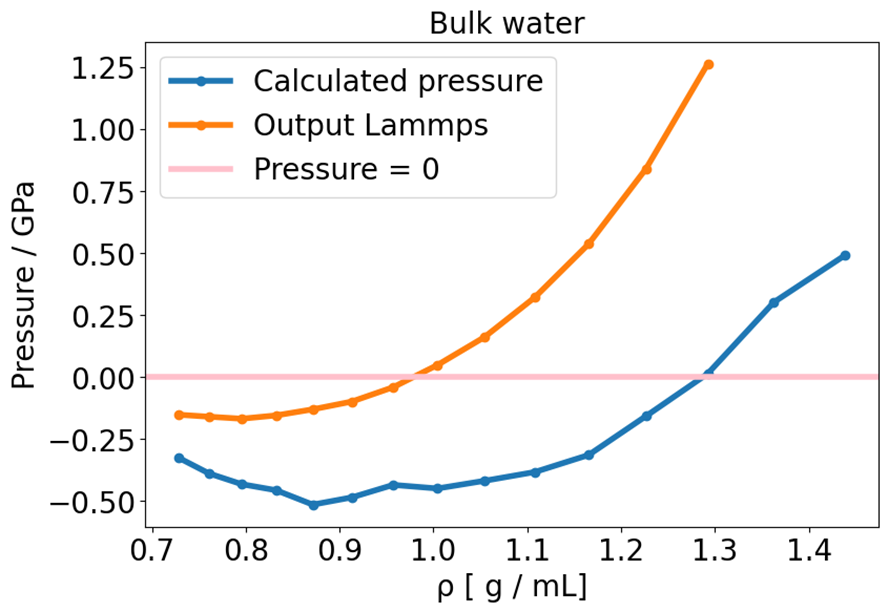
I noticed some inconsistent behavior, where essentially the E-V minimum, located around 1.3 g /mL, does not match of the point where the pressure is zero (around 1.0 g /mL) . There is a mismatch between energy and pressure.

In order to sort this out, I tried out a wide range of possibilities, such as investigating the thermostat (Langevin x CSVR), time constant , time step (0.1-1.0 fs), simulation time (up to 20 ns), number of molecules (up to 1000), pppm cutoff (down to 10^-10 ), coulomb cutoff (10-20 Å ), lammps version, shake x rattle, shake or rattle cutoff (down to 10^-10 ), increased H -mass (1-16 g), using harmonic bonds instead of fixed bonds and tail corrections.
None could solve this E-V mismatch. To compare this with another system, I used NaCl rocksalt and varied the lattice constant. Here there is no discrepancy and the E-V minimum corresponds to where P= 0 .


What made the E-V minimum correspond to where P= 0 for water, was completely removing any kind of bonded interaction, so this behavior is connected to the presence of the bonds, somehow. Of course if I remove the bonds I don’t have a reasonable water model anymore but it suggests that this issue is somehow related to the bonds.
My questions therefore are:
- Is this E-V pressure mismatch inherent to TIP3P or similar bonded-water models? If not, how could I attempt solve it?
- The context of this is that I will add an ion to water in order to calculate a solvation energy. I want to do this by having E_{\text{ion_H2O}} - E_{\text{bulk_H2O}}, both cases at zero pressure. So for this I needed the proper energy at zero pressure, a.k.a the E-V minimum which currently is inconsistent. Since I will use an energy difference in the end, maybe the E-V pressure mismatch is not so relevant?
The version I used was 2021.10.27, however I tried with 2023.08.02 as well and didn’t see any difference.
I attach an example file of a simulation with Rattle.
control.inp (675 Bytes)
structure.inp (11.0 KB)
potential.inp (428 Bytes)