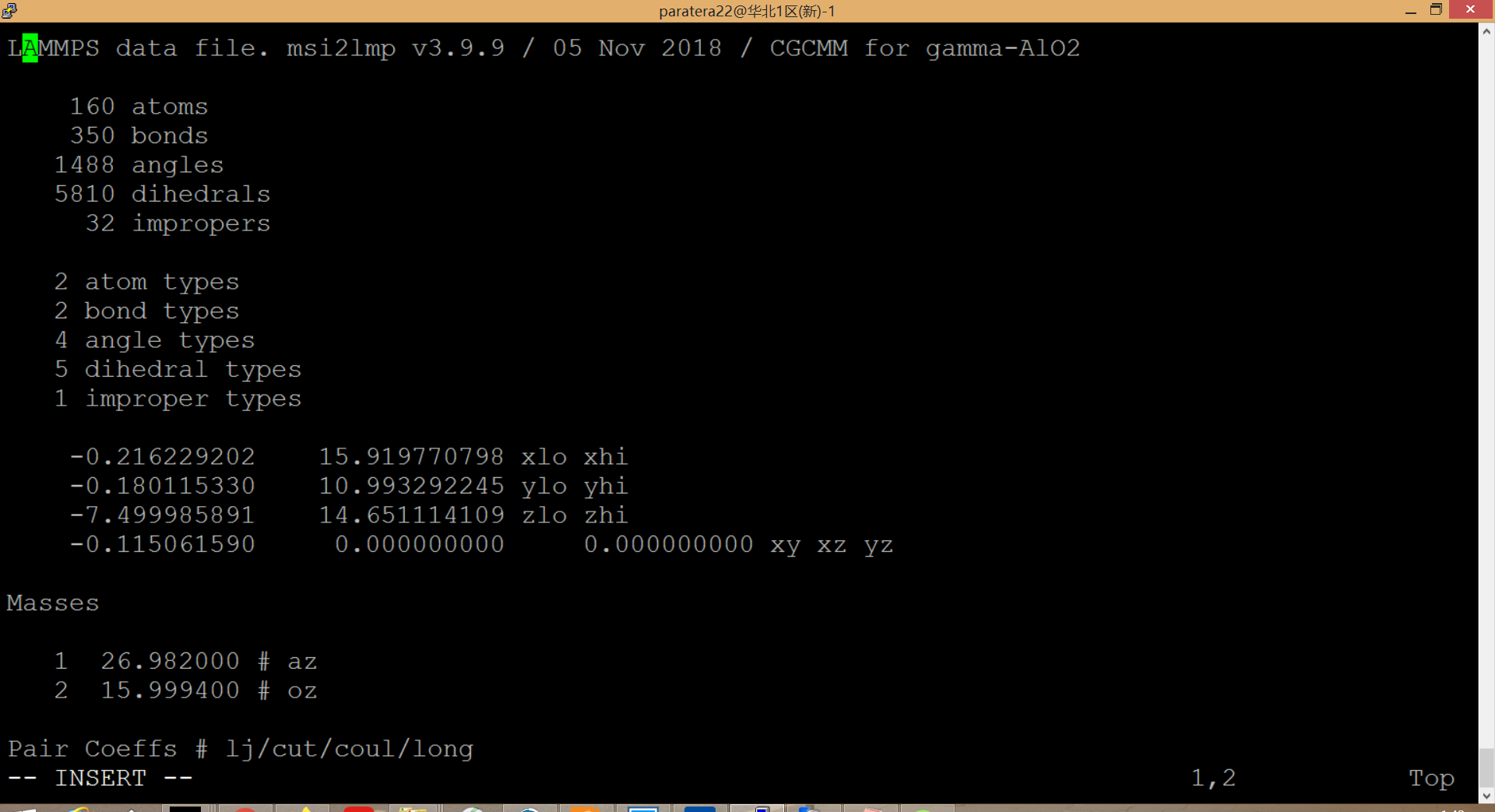
Thanks a lot for your reply. 1) The lammps version is lammps-5Jun19; 2) The platform is Red Hat Enterprise Linux Server release 6.5; 3) The input file is in.PtAlO attached as below; 4) The data file is AlO.data shown as below; 5) The log file is shown as below; 6) The command line used to launch lammps is shown in file sub.sbatch; 7) The original .car and .mdf files are shown below; 8) The command line I use to run msi2lmp: …/msi2lmp.exe gamma-AlO2 -i -frc …/frc_files/cvff.frc. I really appreciate your help.
your input deck is ignoring important information from the documentation. the most important one is: when creating the simulation box, certain system properties are “locked in” and cannot be changed later. those are, for example, the number of atom types, the number of bond/angle/dihedral/improper types, the size of per-atom storage to store bond/angle/dihedral/improper and special neighbor data. this is the reason for the error message you see.
you use the command “create_box 4 total” to define the simulation box. this will reserve space for 4 atom types and NOTHING ELSE.
thus when you use the command “read_data AlO.data add append group AlO shift 52 51 17” it has to error out since it cannot store bond information, since no storage for it has been reserved.
please re-read the documentation of “create_box” and “read_data” very carefully with this in mind and you should be able to resolve this specific problem.
there will be other problems unrelated to that:
your data file is - contrary to your claims - not created by msi2lmp but by LAMMPS and thus contains NO force field parameter assignments.
your data file contains bonds, angles and dihedrals, but there are no bond/angle/dihedral styles defined in your input deck nor are there parameter assignments through the corresponding coeff commands.
your data file was created with an outdated version of topotools based on coordinate data that contains no simulation cell information (its box is set to 1x1x1 angstrom). the latest version (1.8) of topotools will refuse to write data files when the size of the box is not set.
your data file use incorrect atom typing and its numbers are incompatible with your input (there are 136 atom types but your input assumes 4).
in general, you should first figure out how to properly run simulations of each subsystem (i.e. the Pt and the AlO subsystem ) separately before trying for the combined system (and it will be much easier to extend the box and add the Pt to an AlO system instead of the other way around.
it will take significant effort and a much better understanding of the process or how LAMMPS handles and interprets force field data before you will be able to run a successful and meaningful simulation of this kind of system.