`Dear EMC! I got the question below as I try to build a semicrystalline cellulose in version 9.4.4.First,I want to convert crystal file to emc format and then use emc files to Simulation calculations。
Later I also had this problem when I converted the PDB file to EMC format.Urgently need to be solved, thank you!
Dear user,
Your reported error looks very unexpected, which means I can only guess as to what causes it. The root cause clearly is your imported structure. I could check what goes wrong, if you are willing to share both your .car and .mdf file and/or the .pdb and .psf combination you used (.car is preferred, since it represents a richer format).
1 Like
Thank you very much for your reply, since it was a new user who was told that the file could not be uploaded, I have uploaded a screenshot of the contents of the car file below the first chat. AND since new users can only put 3 embedded media items in a post, I put a screenshot of the mdf file here. Looking forward to hearing from you.
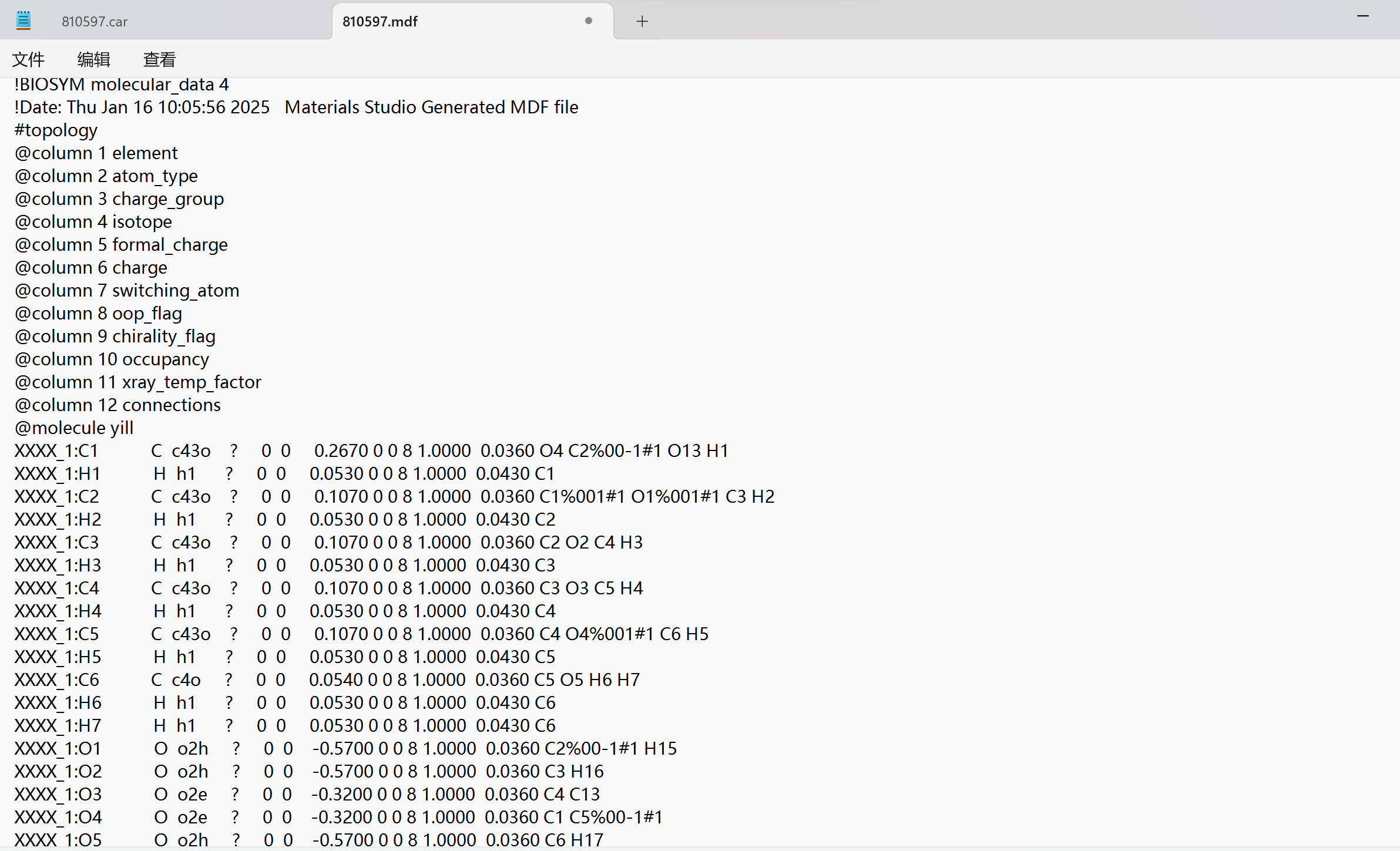
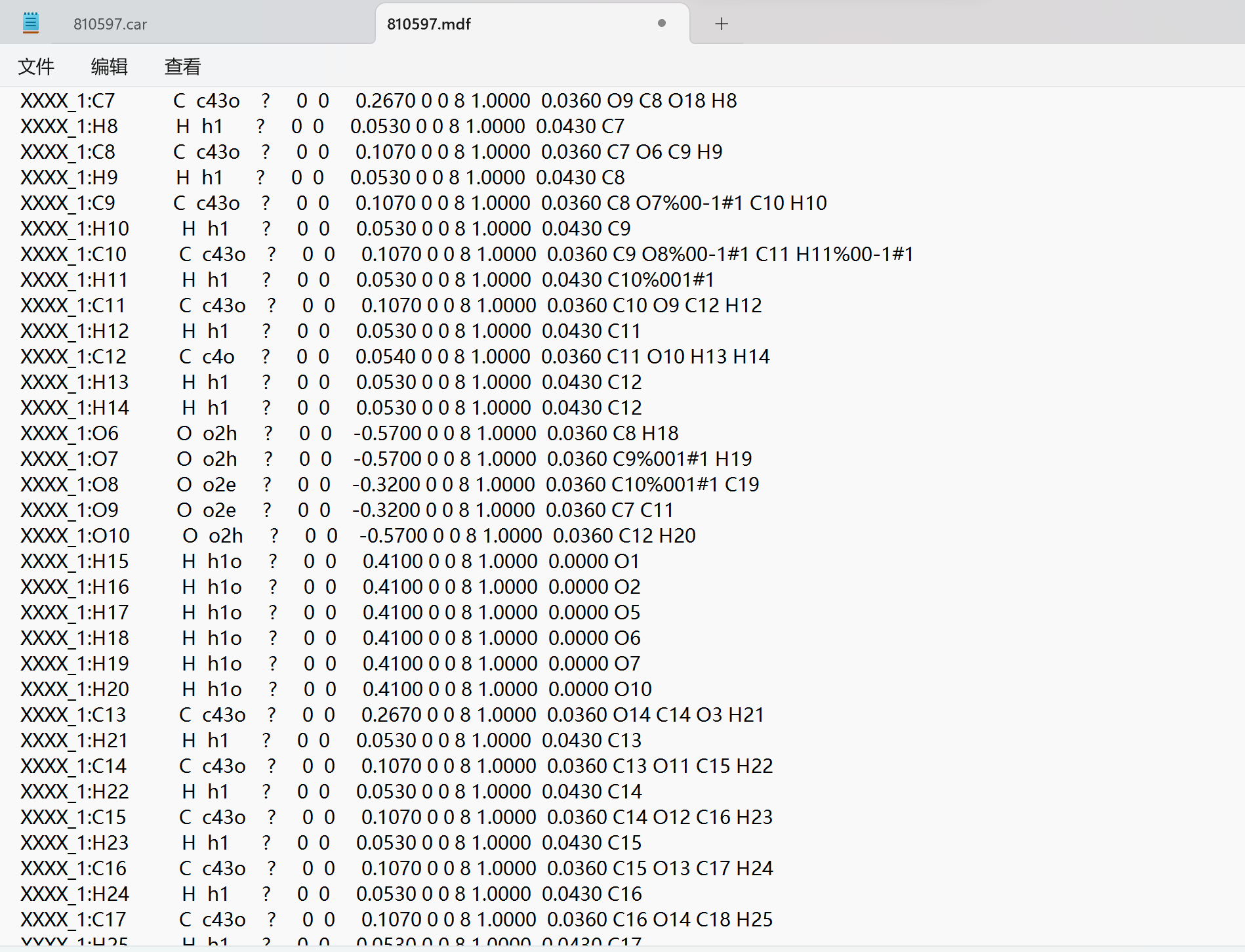
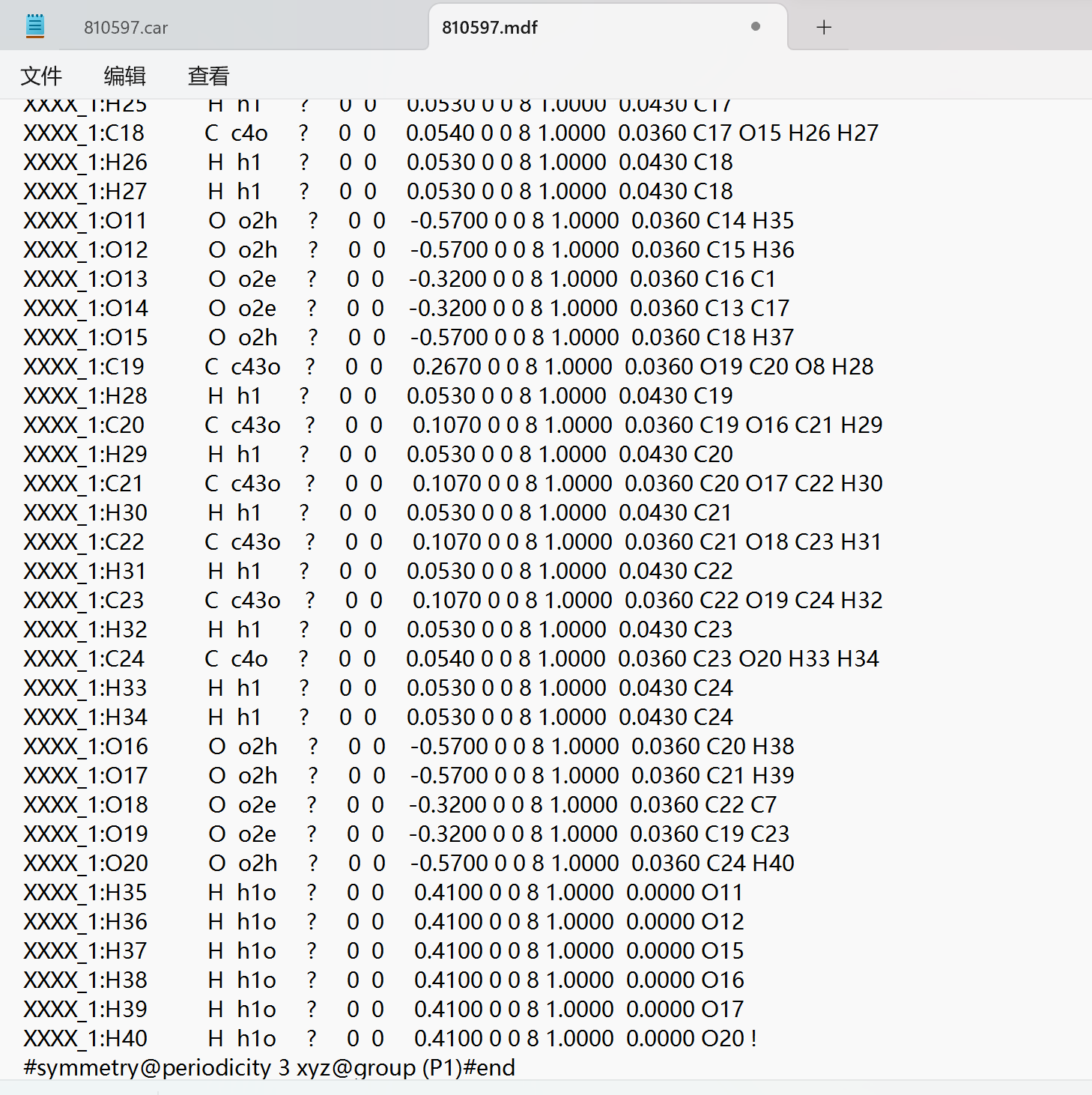
I am terribly sorry, but screen shots are unfortunately not very helpful. You will have to upload your .car etc. as a textual file. Either that or — as a method of last resort, when no other options are available — you could copy paste them as text in your posted message.
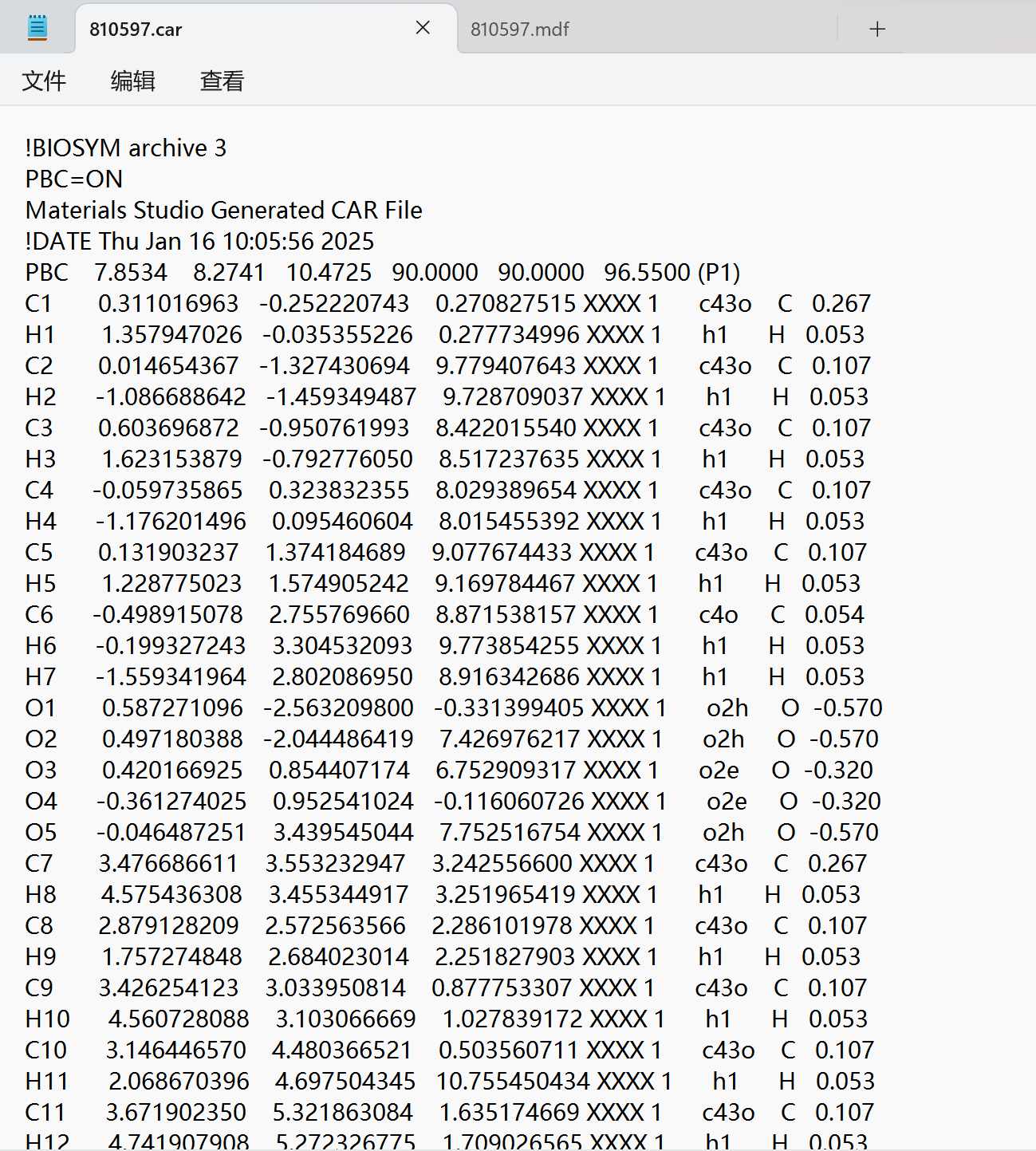
!BIOSYM archive 3
PBC=ON
Materials Studio Generated CAR File
!DATE Thu Jan 16 10:05:56 2025
PBC 7.8534 8.2741 10.4725 90.0000 90.0000 96.5500 (P1)
C1 0.311016963 -0.252220743 0.270827515 XXXX 1 c43o C 0.267
H1 1.357947026 -0.035355226 0.277734996 XXXX 1 h1 H 0.053
C2 0.014654367 -1.327430694 9.779407643 XXXX 1 c43o C 0.107
H2 -1.086688642 -1.459349487 9.728709037 XXXX 1 h1 H 0.053
C3 0.603696872 -0.950761993 8.422015540 XXXX 1 c43o C 0.107
H3 1.623153879 -0.792776050 8.517237635 XXXX 1 h1 H 0.053
C4 -0.059735865 0.323832355 8.029389654 XXXX 1 c43o C 0.107
H4 -1.176201496 0.095460604 8.015455392 XXXX 1 h1 H 0.053
C5 0.131903237 1.374184689 9.077674433 XXXX 1 c43o C 0.107
H5 1.228775023 1.574905242 9.169784467 XXXX 1 h1 H 0.053
C6 -0.498915078 2.755769660 8.871538157 XXXX 1 c4o C 0.054
H6 -0.199327243 3.304532093 9.773854255 XXXX 1 h1 H 0.053
H7 -1.559341964 2.802086950 8.916342686 XXXX 1 h1 H 0.053
O1 0.587271096 -2.563209800 -0.331399405 XXXX 1 o2h O -0.570
O2 0.497180388 -2.044486419 7.426976217 XXXX 1 o2h O -0.570
O3 0.420166925 0.854407174 6.752909317 XXXX 1 o2e O -0.320
O4 -0.361274025 0.952541024 -0.116060726 XXXX 1 o2e O -0.320
O5 -0.046487251 3.439545044 7.752516754 XXXX 1 o2h O -0.570
C7 3.476686611 3.553232947 3.242556600 XXXX 1 c43o C 0.267
H8 4.575436308 3.455344917 3.251965419 XXXX 1 h1 H 0.053
C8 2.879128209 2.572563566 2.286101978 XXXX 1 c43o C 0.107
H9 1.757274848 2.684023014 2.251827903 XXXX 1 h1 H 0.053
C9 3.426254123 3.033950814 0.877753307 XXXX 1 c43o C 0.107
H10 4.560728088 3.103066669 1.027839172 XXXX 1 h1 H 0.053
C10 3.146446570 4.480366521 0.503560711 XXXX 1 c43o C 0.107
H11 2.068670396 4.697504345 10.755450434 XXXX 1 h1 H 0.053
C11 3.671902350 5.321863084 1.635174669 XXXX 1 c43o C 0.107
H12 4.741907908 5.272326775 1.709026565 XXXX 1 h1 H 0.053
C12 3.141965999 6.730797356 1.498262401 XXXX 1 c4o C 0.054
H13 3.411938791 7.273292865 2.383040204 XXXX 1 h1 H 0.053
H14 2.021351005 6.567156939 1.572992165 XXXX 1 h1 H 0.053
O6 3.370567767 1.274827035 2.528281130 XXXX 1 o2h O -0.570
O7 3.193401951 2.068951021 10.305814710 XXXX 1 o2h O -0.570
O8 3.929047125 4.862666572 9.852709128 XXXX 1 o2e O -0.320
O9 3.071358398 4.837747830 2.835102846 XXXX 1 o2e O -0.320
O10 3.516336304 7.569553338 0.425692440 XXXX 1 o2h O -0.570
H15 0.335445381 -2.748883596 0.570300001 XXXX 1 h1o H 0.410
H16 1.081815555 -1.709085578 6.747172848 XXXX 1 h1o H 0.410
H17 0.009395853 4.362575123 8.019996466 XXXX 1 h1o H 0.410
H18 3.233731555 0.849975331 1.658556129 XXXX 1 h1o H 0.410
H19 3.669885104 2.409117587 9.544662833 XXXX 1 h1o H 0.410
H20 3.259631725 7.114047747 -0.391545835 XXXX 1 h1o H 0.410
C13 -0.082621615 0.355914317 5.561863408 XXXX 1 c43o C 0.267
H21 -1.113857900 0.084392394 5.759364174 XXXX 1 h1 H 0.053
C14 -0.045109110 1.503696777 4.641154132 XXXX 1 c43o C 0.107
H22 0.940829841 1.901023172 4.545821844 XXXX 1 h1 H 0.053
C15 -0.585267840 1.067694434 3.297998482 XXXX 1 c43o C 0.107
H23 -1.633879481 0.624469618 3.525611513 XXXX 1 h1 H 0.053
C16 0.257795979 -0.108978699 2.835869577 XXXX 1 c43o C 0.107
H24 1.286163732 0.234062735 2.952548407 XXXX 1 h1 H 0.053
C17 0.161677395 -1.269542628 3.849096939 XXXX 1 c43o C 0.107
H25 -0.934471991 -1.587524200 3.802177329 XXXX 1 h1 H 0.053
C18 1.002009419 -2.509545959 3.643667458 XXXX 1 c4o C 0.054
H26 1.020972677 -3.075128795 4.618563786 XXXX 1 h1 H 0.053
H27 2.060024413 -2.292351555 3.405804207 XXXX 1 h1 H 0.053
O11 -0.776694296 2.536726381 5.285701326 XXXX 1 o2h O -0.570
O12 -0.777535820 2.089581080 2.320835862 XXXX 1 o2h O -0.570
O13 -0.080268864 -0.798354731 1.594225109 XXXX 1 o2e O -0.320
O14 0.640737979 -0.744759062 5.064675977 XXXX 1 o2e O -0.320
O15 0.423044372 -3.433506687 2.703903177 XXXX 1 o2h O -0.570
C19 3.434648006 4.488947352 8.575717803 XXXX 1 c43o C 0.267
H28 2.340831774 4.316723619 8.717945237 XXXX 1 h1 H 0.053
C20 3.620531712 5.538539959 7.469677171 XXXX 1 c43o C 0.107
H29 4.746246277 5.619322581 7.374342179 XXXX 1 h1 H 0.053
C21 2.996761997 5.028557746 6.159210580 XXXX 1 c43o C 0.107
H30 1.899322287 4.796031418 6.484493562 XXXX 1 h1 H 0.053
C22 3.627334941 3.715877762 5.713669879 XXXX 1 c43o C 0.107
H31 4.745597936 3.717649057 5.643967081 XXXX 1 h1 H 0.053
C23 3.373604466 2.793462880 6.932903557 XXXX 1 c43o C 0.107
H32 2.206631096 2.740660149 7.013770028 XXXX 1 h1 H 0.053
C24 3.891272962 1.400740634 6.722598648 XXXX 1 c4o C 0.054
H33 3.751038257 0.949179303 7.667389597 XXXX 1 h1 H 0.053
H34 5.020583843 1.442715388 6.687657504 XXXX 1 h1 H 0.053
O16 3.061932901 6.836125556 7.872432520 XXXX 1 o2h O -0.570
O17 3.187102197 6.012984256 5.217329118 XXXX 1 o2h O -0.570
O18 2.949186500 3.190076722 4.564328662 XXXX 1 o2e O -0.320
O19 4.015964080 3.268654553 8.124875849 XXXX 1 o2e O -0.320
O20 3.348457252 0.669861773 5.671993552 XXXX 1 o2h O -0.570
H35 -0.296679683 2.600079760 6.133316690 XXXX 1 h1o H 0.410
H36 -1.148198465 1.580191452 1.594981053 XXXX 1 h1o H 0.410
H37 0.445098606 -4.284623147 3.167660956 XXXX 1 h1o H 0.410
H38 3.101921677 7.368455485 7.064795073 XXXX 1 h1o H 0.410
H39 2.781760308 5.634731029 4.430415238 XXXX 1 h1o H 0.410
H40 3.719369842 0.927289396 4.834620142 XXXX 1 h1o H 0.410
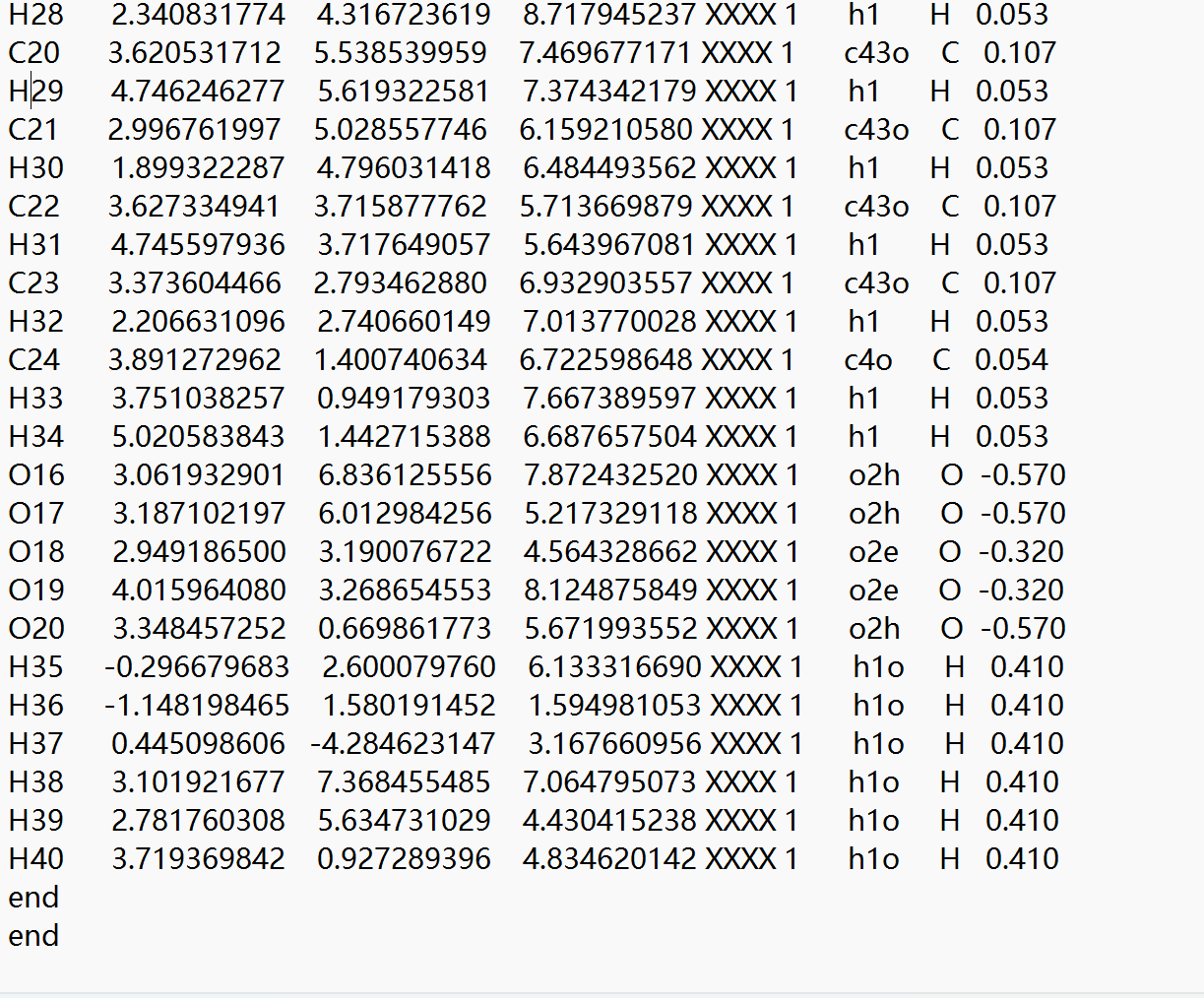
end
end
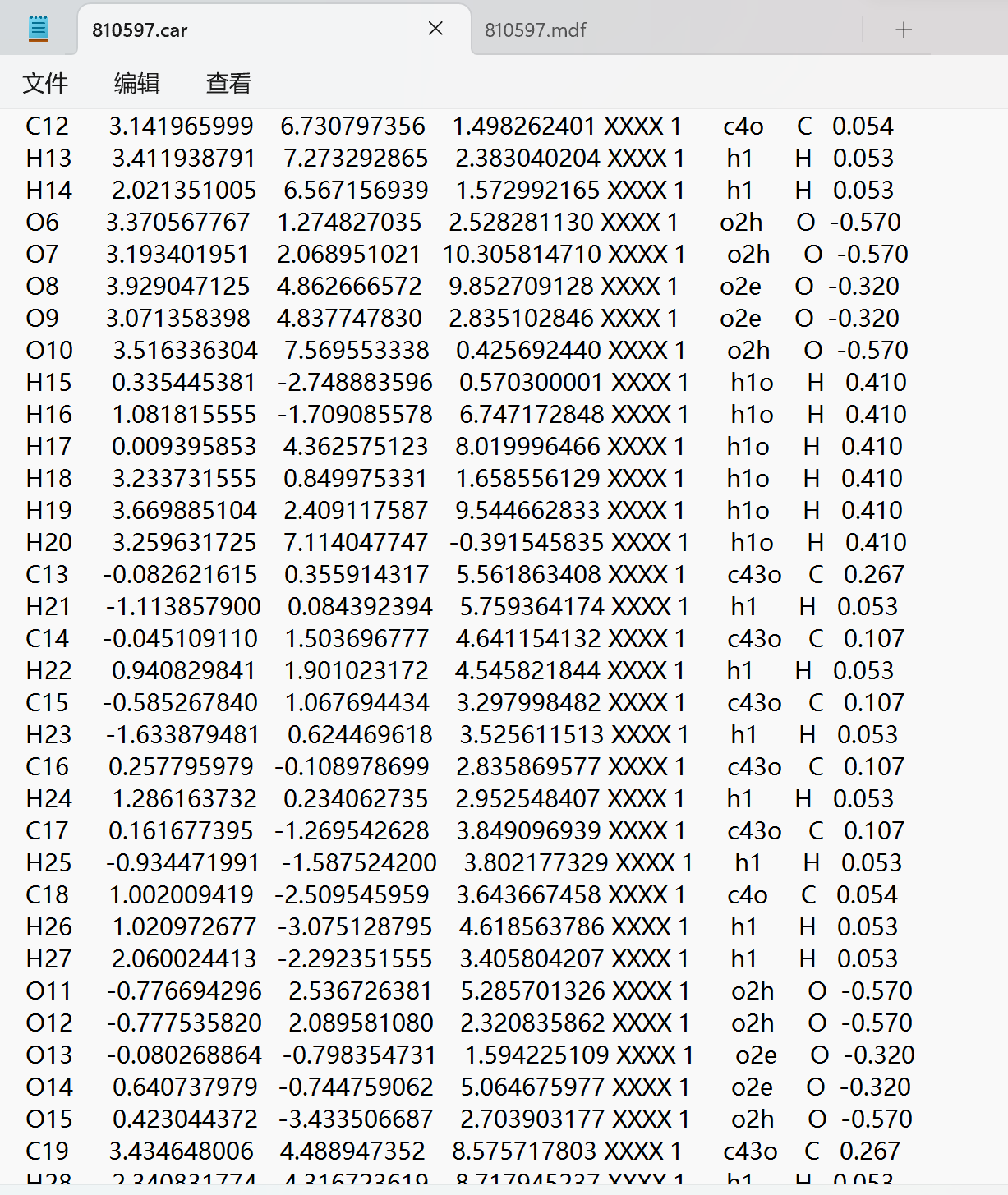
The content of my car file is the same as the screenshot.,If you don’t have anything to do.,Can you give me other suggestions?。 I have a great need for EMC to build semi-crystalline substances. The basic unit of crystal structure is the Iβ unit cell. I have modeled a semi-crystalline polyethylene method based on an example in the software.
Could you provide your .mdf and .esh as well? Thanks!
This is my mdf file.
!BIOSYM molecular_data 4
!Date: Thu Jan 16 10:05:56 2025 Materials Studio Generated MDF file
@column 1 element
@column 2 atom_type
@column 3 charge_group
@column 4 isotope
@column 5 formal_charge
@column 6 charge
@column 7 switching_atom
@column 8 oop_flag
@column 9 chirality_flag
@column 10 occupancy
@column 11 xray_temp_factor
@column 12 connections
@molecule yill
XXXX_1:C1 C c43o ? 0 0 0.2670 0 0 8 1.0000 0.0360 O4 C2%00-1#1 O13 H1
XXXX_1:H1 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C1
XXXX_1:C2 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C1%001#1 O1%001#1 C3 H2
XXXX_1:H2 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C2
XXXX_1:C3 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C2 O2 C4 H3
XXXX_1:H3 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C3
XXXX_1:C4 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C3 O3 C5 H4
XXXX_1:H4 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C4
XXXX_1:C5 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C4 O4%001#1 C6 H5
XXXX_1:H5 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C5
XXXX_1:C6 C c4o ? 0 0 0.0540 0 0 8 1.0000 0.0360 C5 O5 H6 H7
XXXX_1:H6 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C6
XXXX_1:H7 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C6
XXXX_1:O1 O o2h ? 0 0 -0.5700 0 0 8 1.0000 0.0360 C2%00-1#1 H15
XXXX_1:O2 O o2h ? 0 0 -0.5700 0 0 8 1.0000 0.0360 C3 H16
XXXX_1:O3 O o2e ? 0 0 -0.3200 0 0 8 1.0000 0.0360 C4 C13
XXXX_1:O4 O o2e ? 0 0 -0.3200 0 0 8 1.0000 0.0360 C1 C5%00-1#1
XXXX_1:O5 O o2h ? 0 0 -0.5700 0 0 8 1.0000 0.0360 C6 H17
XXXX_1:C7 C c43o ? 0 0 0.2670 0 0 8 1.0000 0.0360 O9 C8 O18 H8
XXXX_1:H8 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C7
XXXX_1:C8 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C7 O6 C9 H9
XXXX_1:H9 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C8
XXXX_1:C9 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C8 O7%00-1#1 C10 H10
XXXX_1:H10 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C9
XXXX_1:C10 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C9 O8%00-1#1 C11 H11%00-1#1
XXXX_1:H11 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C10%001#1
XXXX_1:C11 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C10 O9 C12 H12
XXXX_1:H12 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C11
XXXX_1:C12 C c4o ? 0 0 0.0540 0 0 8 1.0000 0.0360 C11 O10 H13 H14
XXXX_1:H13 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C12
XXXX_1:H14 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C12
XXXX_1:O6 O o2h ? 0 0 -0.5700 0 0 8 1.0000 0.0360 C8 H18
XXXX_1:O7 O o2h ? 0 0 -0.5700 0 0 8 1.0000 0.0360 C9%001#1 H19
XXXX_1:O8 O o2e ? 0 0 -0.3200 0 0 8 1.0000 0.0360 C10%001#1 C19
XXXX_1:O9 O o2e ? 0 0 -0.3200 0 0 8 1.0000 0.0360 C7 C11
XXXX_1:O10 O o2h ? 0 0 -0.5700 0 0 8 1.0000 0.0360 C12 H20
XXXX_1:H15 H h1o ? 0 0 0.4100 0 0 8 1.0000 0.0000 O1
XXXX_1:H16 H h1o ? 0 0 0.4100 0 0 8 1.0000 0.0000 O2
XXXX_1:H17 H h1o ? 0 0 0.4100 0 0 8 1.0000 0.0000 O5
XXXX_1:H18 H h1o ? 0 0 0.4100 0 0 8 1.0000 0.0000 O6
XXXX_1:H19 H h1o ? 0 0 0.4100 0 0 8 1.0000 0.0000 O7
XXXX_1:H20 H h1o ? 0 0 0.4100 0 0 8 1.0000 0.0000 O10
XXXX_1:C13 C c43o ? 0 0 0.2670 0 0 8 1.0000 0.0360 O14 C14 O3 H21
XXXX_1:H21 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C13
XXXX_1:C14 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C13 O11 C15 H22
XXXX_1:H22 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C14
XXXX_1:C15 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C14 O12 C16 H23
XXXX_1:H23 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C15
XXXX_1:C16 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C15 O13 C17 H24
XXXX_1:H24 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C16
XXXX_1:C17 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C16 O14 C18 H25
XXXX_1:H25 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C17
XXXX_1:C18 C c4o ? 0 0 0.0540 0 0 8 1.0000 0.0360 C17 O15 H26 H27
XXXX_1:H26 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C18
XXXX_1:H27 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C18
XXXX_1:O11 O o2h ? 0 0 -0.5700 0 0 8 1.0000 0.0360 C14 H35
XXXX_1:O12 O o2h ? 0 0 -0.5700 0 0 8 1.0000 0.0360 C15 H36
XXXX_1:O13 O o2e ? 0 0 -0.3200 0 0 8 1.0000 0.0360 C16 C1
XXXX_1:O14 O o2e ? 0 0 -0.3200 0 0 8 1.0000 0.0360 C13 C17
XXXX_1:O15 O o2h ? 0 0 -0.5700 0 0 8 1.0000 0.0360 C18 H37
XXXX_1:C19 C c43o ? 0 0 0.2670 0 0 8 1.0000 0.0360 O19 C20 O8 H28
XXXX_1:H28 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C19
XXXX_1:C20 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C19 O16 C21 H29
XXXX_1:H29 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C20
XXXX_1:C21 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C20 O17 C22 H30
XXXX_1:H30 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C21
XXXX_1:C22 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C21 O18 C23 H31
XXXX_1:H31 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C22
XXXX_1:C23 C c43o ? 0 0 0.1070 0 0 8 1.0000 0.0360 C22 O19 C24 H32
XXXX_1:H32 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C23
XXXX_1:C24 C c4o ? 0 0 0.0540 0 0 8 1.0000 0.0360 C23 O20 H33 H34
XXXX_1:H33 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C24
XXXX_1:H34 H h1 ? 0 0 0.0530 0 0 8 1.0000 0.0430 C24
XXXX_1:O16 O o2h ? 0 0 -0.5700 0 0 8 1.0000 0.0360 C20 H38
XXXX_1:O17 O o2h ? 0 0 -0.5700 0 0 8 1.0000 0.0360 C21 H39
XXXX_1:O18 O o2e ? 0 0 -0.3200 0 0 8 1.0000 0.0360 C22 C7
XXXX_1:O19 O o2e ? 0 0 -0.3200 0 0 8 1.0000 0.0360 C19 C23
XXXX_1:O20 O o2h ? 0 0 -0.5700 0 0 8 1.0000 0.0360 C24 H40
XXXX_1:H35 H h1o ? 0 0 0.4100 0 0 8 1.0000 0.0000 O11
XXXX_1:H36 H h1o ? 0 0 0.4100 0 0 8 1.0000 0.0000 O12
XXXX_1:H37 H h1o ? 0 0 0.4100 0 0 8 1.0000 0.0000 O15
XXXX_1:H38 H h1o ? 0 0 0.4100 0 0 8 1.0000 0.0000 O16
XXXX_1:H39 H h1o ? 0 0 0.4100 0 0 8 1.0000 0.0000 O17
XXXX_1:H40 H h1o ? 0 0 0.4100 0 0 8 1.0000 0.0000 O20
!
#symmetry
@ periodicity 3 xyz
@ group (P1)
#end
This is my convert.emc document and the error message is placed at the end
(* EMC: Script *)
(* 定义变量 *)
variables = {
input → “810597geo.car”,
output → “cellulose.emc”
};
(* 定义类型 *)
types = {
atomistic → true
};
(* 定义系统 )
systems = {
n → 1,
properties → {
id → 0,
p → 1.0,
moves → false ( 禁用分子运动 *)
}
};
(* 导入CAR文件 )
insight = {
name → input,
forcefield → “standard”,
system → 0,
fixed → true ( 固定原子位置 *)
};
(* 初始化力场 *)
force = {
style → “init”,
message → true
};
(* 输出EMC文件 *)
put = {
name → output
};
CODE run process
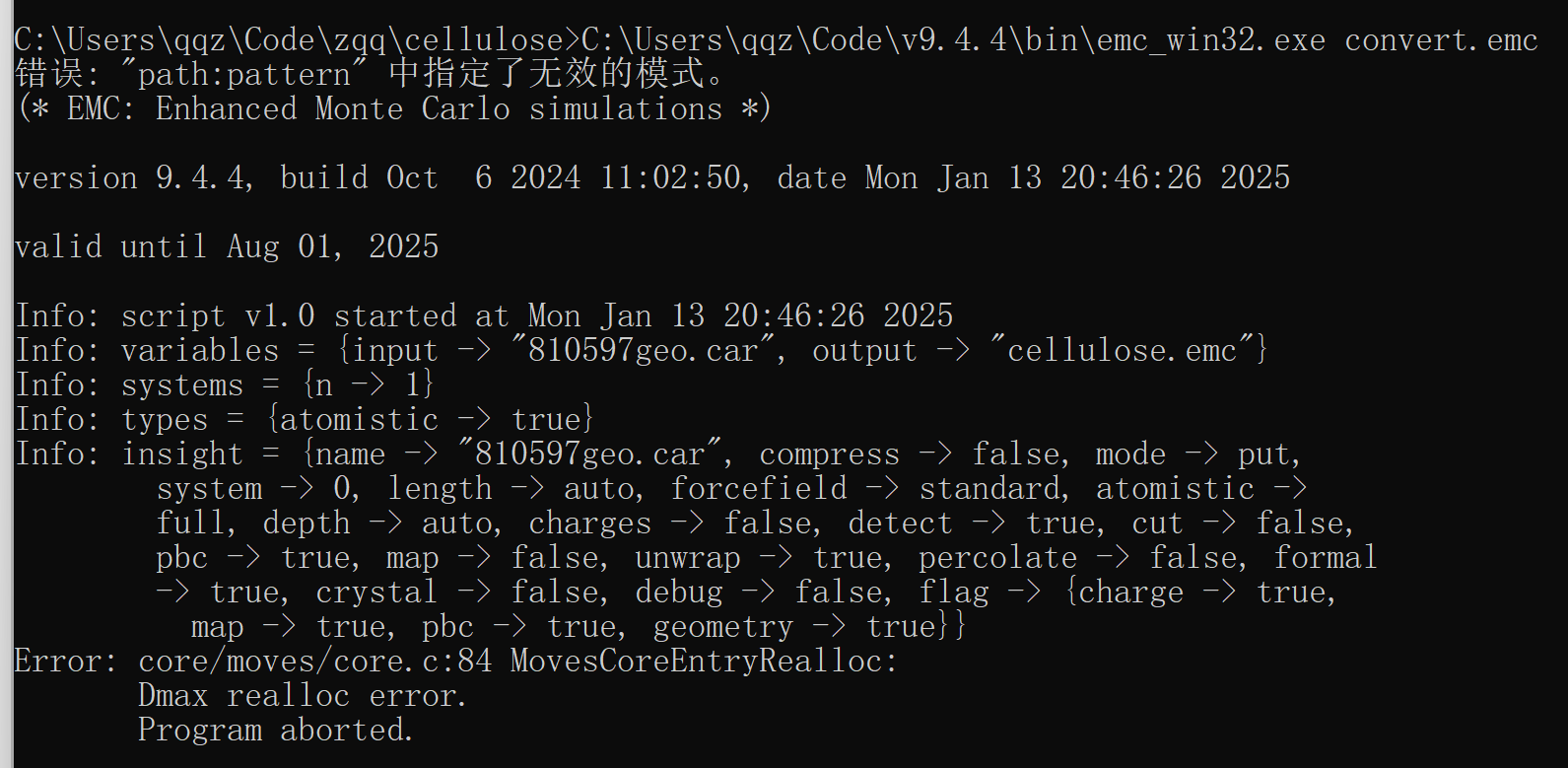
C:\Users\qqz\Code\zqq\cellulose>C:\Users\qqz\Code\v9.4.4\bin\emc_win32.exe convert.emc
错误: “path:pattern” 中指定了无效的模式。
(* EMC: Enhanced Monte Carlo simulations *)
version 9.4.4, build Oct 6 2024 11:02:50, date Fri Jan 17 16:30:50 2025
valid until Aug 01, 2025
Info: script v1.0 started at Fri Jan 17 16:30:50 2025
Info: variables = {input → “810597geo.car”, output → “cellulose.emc”}
Info: types = {atomistic → true}
Info: systems = {n → 1}
Info: insight = {name → “810597geo.car”, compress → false, mode → put,
system → 0, length → auto, forcefield → standard, atomistic →
full, depth → auto, charges → false, detect → true, cut → false,
pbc → false, map → false, unwrap → true, percolate → false, formal
→ true, crystal → false, debug → false, flag → {charge → true,
map → true, pbc → true, geometry → true}}
Error: core/moves/core.c:84 MovesCoreEntryRealloc:
Dmax realloc error.
Program aborted.
Can you see what’s wrong,thanks
Dear user,
I checked out your reported issue, but I could not reproduce the reported error. First of all, I would suggest using an .esh file instead of an .emc file to set up your simulation:
# Options section
ITEM OPTIONS
replace true
field charmm/c36a/cgenff
number true
ntotal 10
density 0.1
emc_execute true
ITEM END # OPTIONS
# Clusters section
ITEM CLUSTERS
molecule import, name="cellulose.car", &
type=crystal, percolate=true, depth=8
ITEM END # CLUSTERS
This generates build.emc, which can be used for the correct importing approach. Main issue in your .emc is that you forgot to set the force field before loading your crystal structure. The latter structure, however, suffers from some issues. EMC tries to create groups based on the provided structure, guessing rings etc. Unfortunately – due to the fact that your sugar rings cross box boundaries – EMC is currently not capable of deriving correct group representations, thus hampering subsequent structure interpretation.
Apart from these technical issues, your premise to use EMC to build semicrystalline input structures will not work, since the semicrystalline approach currently only works for linear united atom chains without any decorations or rings.