Hello .
I need several interatomic interactions. In this regard, I want to use 3 reax potentials that are made for one subject. But when I use the following command, I get an error
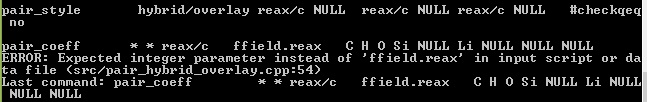
pair_style hybrid/overlay reax/c NULL reax/c NULL reax/c NULL #checkqeq no
pair_coeff * * reax/c ffield.reax C H O Si NULL Li NULL NULL NULL
pair_coeff * * reax/c ffield1.reax NULL H O Si Al Li NULL NULL NULL
pair_coeff * * reax/c ffield2.reax C NULL NULL NULL Al NULL NULL NULL NULL
Another error occurs when I change the command to below.
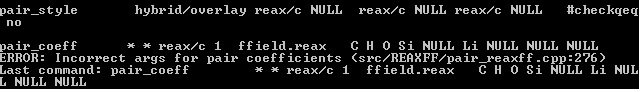
pair_style hybrid/overlay reax/c NULL reax/c NULL reax/c NULL #checkqeq no
pair_coeff * * reax/c 1 ffield.reax C H O Si NULL Li NULL NULL NULL
pair_coeff * * reax/c 2 ffield1.reax NULL H O Si Al Li NULL NULL NULL
pair_coeff * * reax/c 3 ffield2.reax C NULL NULL NULL Al NULL NULL NULL NULL
Is it correct that I have used several reax potentials or is this wrong ?
How to solve these error ?
What you are trying to do is bad or wrong in about every aspect you look at it
In general ReaxFF is not a force field that lends itself well to hybrid pair styles. It is intended to be used without a hybrid pair style and a single parameter file
If at all, the correct style would be hybrid and not hybrid/overlay. The latter will double count interactions mentioned in multiple pair styles. hybrid/overlay would be useful to add coulomb to a pair style that does not include coulomb (i.e. hybrid/overlay lj/cut 12.0 coul/long 12.0 would be the same as lj/cut/coul/long 12.0).
When using hybrid pair styles like ReaxFF that require a single pair_coeff command, there must not be any elements shared between different pair_coeff styles, and interactions between the subgroups must be implemented using a pairwise additive potential
Only the second quoted example is syntactically correct, but the error indicates that there is a mismatch between your system and your input. Probably a different number of atom types
There are atom types no assigned any interaction.
When using ReaxFF multiple times, there is no consistent way possible to compute the (required) charge equilibration.
In summary, even if you figure out a way to make LAMMPS accept the syntax of your input file, it will still be a bogus simulation with unphysical results. You need to find a ReaxFF parameter file that is meant to represent your entire system, it will not work to try and stitch together multiple ones.
Which means you have three choices: either find a tutor that will teach you the skills you are lacking, or find a collaborator that will contribute to your work, or find a different research project where the missing kind of skill is not needed.