Hello Everyone,
I have been attempting to characterize polyethylene using the REAX/c implementation of the forcefield and believe I may have detected an error. My system is a single periodic polyethylene chain. I have been attempting to obtain the minimized configuration at 0K. Using box/relax and conjugate gradient gave very poor results with the non-conservative REAX so I wrote my own code to manually find the equilibrium box size with the change_box command and the fire minimizer.
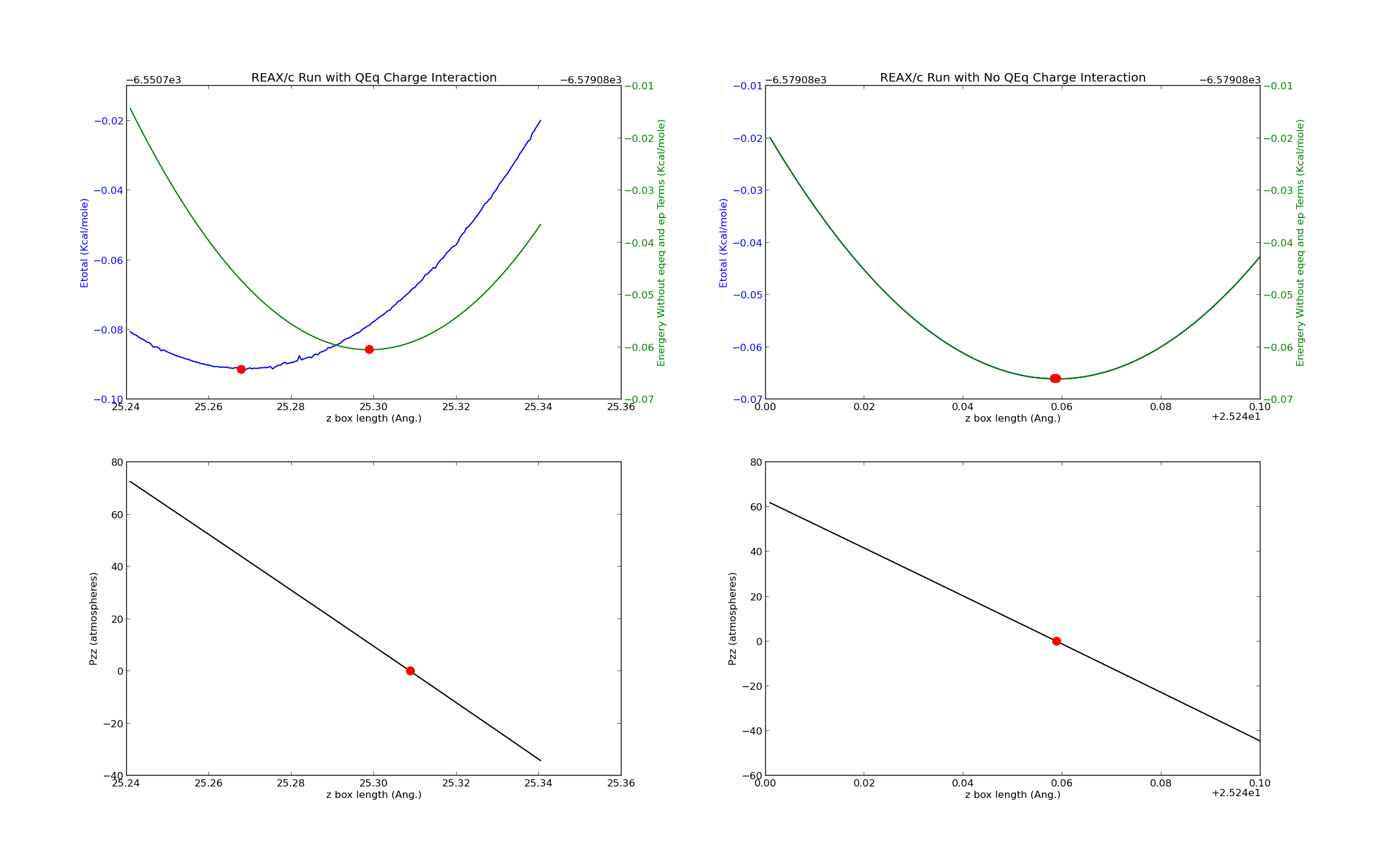
Essentially I adjust the box and minimize the system a certain number of times over an interval and then look at the total energy as a function of that box dimension length (in this case lz) . This has worked well but it provided me with some unusual results:
When using REAX/c with the QEq charge interactions the the minimum in the total energy did not occur at 0 pressure, as it should have. Closer analysis of the individual energy terms generated by REAX showed that removing the coulomb energy (ep) and charge equilibration energy (eqeq) from the total led to an energy minimum at a much closer lz to the one at which pzz ~ 0.
To further investigate I reran my simulation without the QEq charge interactions. In this case the ep and eqeq energy terms are 0 and have no effect (as expected) and the lz of the energy minimum coincides precisely with that of pzz ~ 0.
I have plotted what I just described in a figure I have attached. I have also included on archive of the working directory with the relevant simulation files.
I believe this might point to a failure of REAX/c to properly calculate pressure contributions due to the charge interaction energy terms. I don't have the standard REAX package compiled at the moment so I haven't checked if it also shows this behaviour but it is probably worth looking into.
I haven't dug around in the source code much yet. Honestly I'm not all that knowledgeable about the implementation at that level; but I thought it would be worth bringing this to the attention of folks better equipped than I.
Regards,
Thomas O'Connor
Johns Hopkins University
Dept. of Physics and Astronomy
PE_test.tar.gz (153 KB)