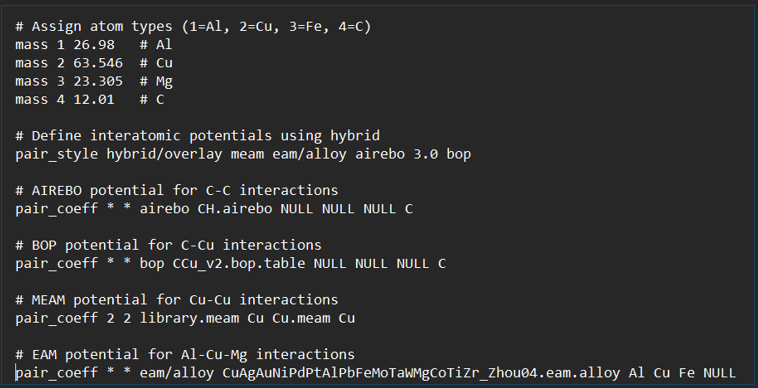
That is incorrect. You should be using pair style hybrid. The hybrid/overlay style is for adding potentials for the same atom types, but in your case that would be double counting interactions.
Apart from the syntax errors you have, there is a conceptual problem. You are treating manybody potentials as if they were pair-wise additive, but they are not. So a split with different pair styles for C-C, C-Cu, and Cu-Cu interactions cannot work for the C-Cu interaction. If you have a many-body potential it not only computes interactions for pairs of atoms, but also for triples. So for C-Cu, you need to look at C-C-Cu, C-Cu-Cu, Cu-C-Cu, Cu-C-C, Cu-Cu-C, C-Cu-C interactions to get them correctly and that can only happen if all interactions between C and Cu are covered by the same pair style, e.g.
pair_coeff * * bop CCu_v2.bop.table NULL Cu NULL C
As you have it, because of using hybrid/overlay you will compute C-C interaction with airebo and add to that the C-C interaction from bop. If you would be using pair style hybrid, then the airebo part would be wiped out by the bop setting.
Another conceptual problem is when using EAM or MEAM. The embedding term is not correctly computed when using a hybrid style since it only considers the atom types within the type assignment and the contributions from atom types assigned to other pair styles is ignored. This also applies to MEAM and EAM in the same hybrid style.
So what does make sense? Actually, very little. Ideally, you would want to use a single pair style for all elements. That would be possible with MEAM, but I am not certain there is a parameterization that has been tuned specifically for your use case, and not having “tuned” parameters with MEAM will incur significant errors in your model, same as for any other force field where you only have “generic” parameters. If the geometry of the nanotube doesn’t matter to you, you can just use fix setforce to freeze its geometry.
If you want to use AIREBO for the nanotube (which only makes sense if you do care significantly about how the nanotube geometry reacts to the surrounding metals and how you manipulate the whole system), you have the challenge of finding a suitable pairwise additive potential parameter set (e.g. lj/cut or morse) for the C- interactions. Then you could use EAM or MEAM for all the metals, but with the caveat of incorrect embedding term for the C- interaction.
Bottom line, while using pair style hybrid may look like the solution to your problems, force fields are not like lego bricks that you can put together in all kinds of ways. Each have their own restrictions and accuracy issues. As a beginner, my recommendation is to find a project that does not require skills and a level of understanding that you currently don’t posses and find a tutor that can teach you these skills. This is something that can take months to years to understand sufficiently well. The art of force field parameterization has a lot of “black magic” in it that is not obvious and easy to learn.