Dear LAMMPS Community,
I am simulating a single particle using the Langevin equation and have encountered an issue I hope you can help with. My workflow involves running LAMMPS (version 29 Aug 2024) in a loop, where I,
1. Run a LAMMPS simulation for a certain number of timesteps.
2. Save the restart file.
3. Use this restart file to continue the simulation for the next loop iteration.
4. Repeat this process for 100,000 loops.
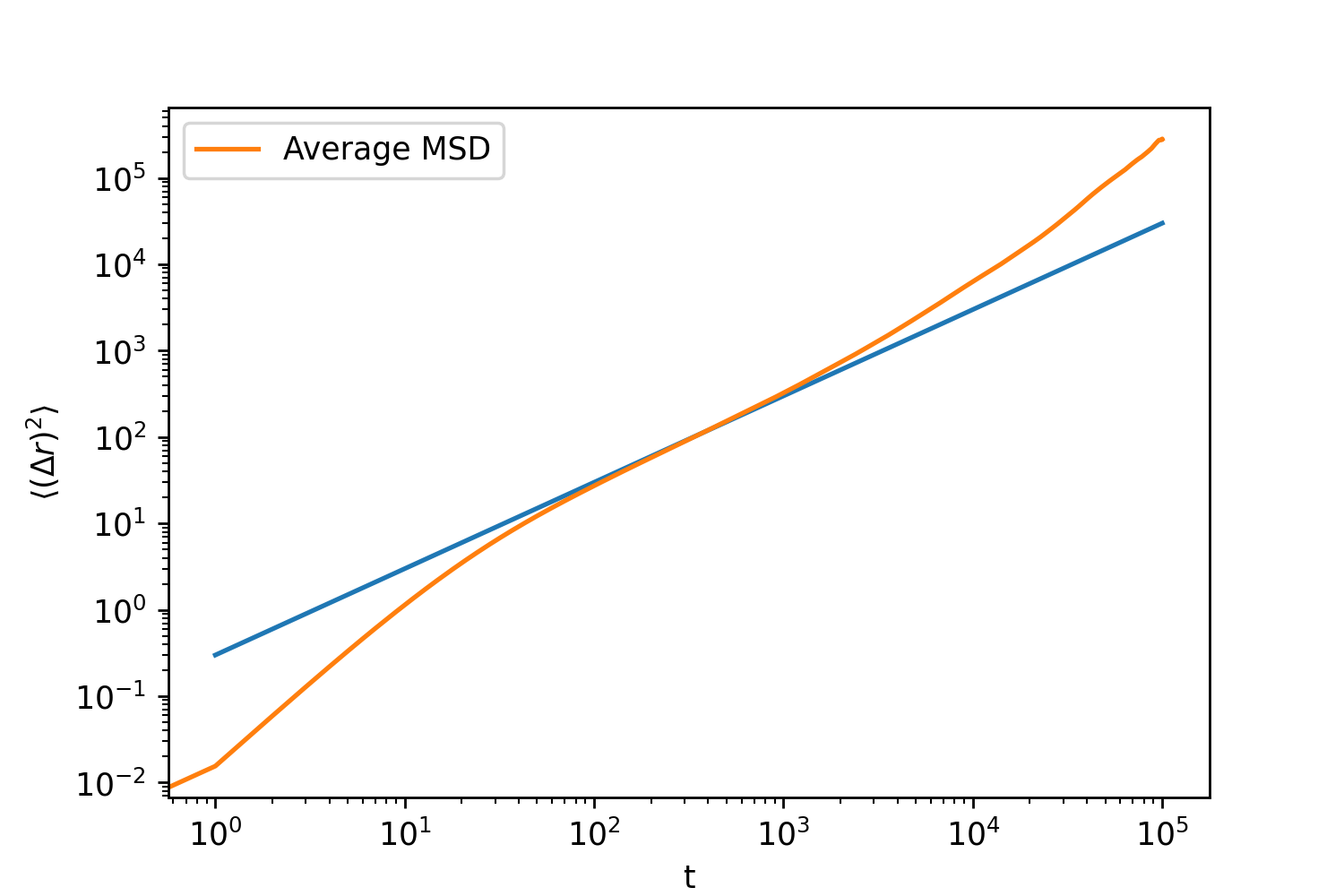
When I plot the mean squared displacement (MSD) as a function of time (t), I observe superdiffusive behavior at longer time, which is unexpected for this setup—I should be seeing normal diffusive behavior.
Interestingly, when I run LAMMPS continuously (without using loops and restart files), I observe the expected diffusive behavior.
I have checked my simulation parameters carefully and ensured consistency between the looped and continuous setups, but the discrepancy persists. Could the use of restart files in a looped setup introduce subtle errors, or is there something else I might be overlooking?
Thank you in advance for your help!
Here is the input script (LAMMPS version 29 Aug 2024):
############################################################
############################################################
Box and units
units lj
atom_style atomic
boundary p p p
READ “start” data file
read_restart restart.restart
log log.txt append
pair_style lj/cut 1.12246152962189
pair_coeff 1 1 1.0 1.0 1.12246152962189
variable seed equal 70
fix 1 all nve # NVE integrator
fix 2 all langevin 1.0 1.0 1.0 ${seed} # langevin thermostat
Output thermodynamic info (temperature, energy, pressure, etc.)##########################
thermo 10000
thermo_style custom step temp etotal pe ke epair emol press vol
#############################################################################################
set timestep of integrator
timestep 0.1
dump 1 all custom 1 dump.position.txt id type xu yu zu
run 1
write_restart restart.restart
#######################