I am currently attempting to run a simulation involving the bombardment of a copper slab with oxygen atoms. I have a deposition region right above the slab where I deposit oxygen atoms with an initial velocity. Atoms that bounce off the slab are evaporated after exceeding the deposition region. I am running into an issue where atoms sometimes hit the surface bounce off with little to no velocity. These atoms float away slowly and spend a long time in the deposition region (they don’t reach the evaporation region) without any neighbors. I speculate that this is causing the Lost Atoms error. I tested it by just depositing atoms with no initial velocity in the deposition zone and it throws the Lost Atoms error after an atom exists without neighbors for too long. It always shows one atom is lost. Ex: orig 3007 curr 3006
My questions are:
Can an atom existing without neighbors for too long cause a Lost Atoms error?
If so, is there a way to have the atom removed instead of throwing an error or prevent the atoms from being lost?
I would also like to save increasing the neighbor list cutoff range as a last resort because the simulation may slow down too much. I am a new user to any advice would be greatly appreciated!
No, atoms existing without neighbors will not cause lost atoms. Lost atoms can happen when two atoms overlap strongly then blow apart, if an atom wanders out of a non-periodic boundary, if your timestep is too large, or if your reneighboring criteria are too aggressive and you are getting “Dangerous builds” which miss interactions.
A more common setup for this kind of simulation is to not remove atoms explicitly, but to have an open box (i.e. a non-periodic, fixed boundary without a wall) and then tell LAMMPS to ignore the atoms that leave the box.
Hi stamoor,
Thanks for the reply. I am currently using all periodic boundaries (p p p) for the simulation. I have a slab of 3000 atoms, 15x20 with a variable timestep:
fix_dt substrate_g dt/reset 10 0.05 1.00 0.01
with reneighboring criteria:
neigh_modify every 10 delay 0 check no exclude group frozen_g frozen_g
I would like to attach my input script, but I don’t seem to be able to as a new user.
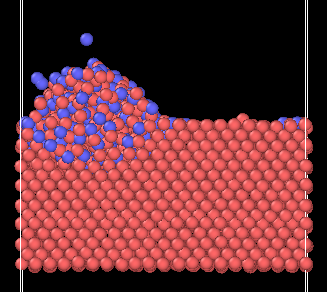
I do have wall/regions with LJ interactions involved in the simulation that could be causing the issue, but I was surprised that depositing atoms without initial velocity was still causing lost atoms. Here is an image of the setup I am trying to achieve:
where the yellow lines are the boundaries for the barriers. This image is a snapshot of a case where I was adding atoms with velocity.
I see. That makes sense, so I don’t have to have a evaporation region that is constantly checking for atoms that have entered. I’ll look into implementing that.
Hi stamoor,
I got the results back, and it still has lost the atom at the exact same timestep as in previous runs.
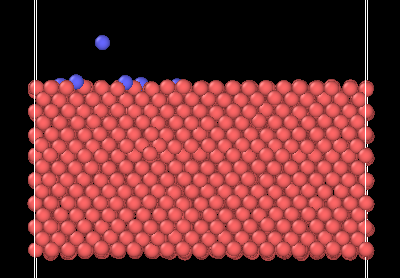
Here are the last snapshots of the structure before it loses an atom (snapshots collected every 100 timesteps):
With initial velocity @ 901300 timesteps
With some with initial velocity, and some without initial velocity @ 12500 timesteps
Both have wall/region LJ.
This error only started happening after adding in the walls. I just had a new thought that it may be because the new atoms are depositing too close to the walls since the edge of the deposition region and the walls were only 0.1 A apart. I’ll try increasing the distance to 2A and see if that is what is causing the issue.