Hello!!! Giving a bit of context, what I want to achieve with this code
(in.eam - Google Drive, in.eam (1.5 KB)) is to collide a copper nanoparticle with a nickel surface (I obtained these files from Ovito, so I’m using read_data Cu.data (18.1 KB), Ni.data - Google Drive )). However, I have an error and I would like to consult it with you:
The code reads the surface or the copper nanoparticle separately (when using #), but it doesn’t read them together. When I try to execute the code with both, I get the following output:
LAMMPS (7 Aug 2019)
OMP_NUM_THREADS environment is not set. Defaulting to 1 thread. (…/comm.cpp:93)
using 1 OpenMP thread(s) per MPI task
OMP_NUM_THREADS environment is not set. Defaulting to 1 thread. (…/comm.cpp:93)
using 1 OpenMP thread(s) per MPI task
Reading data file …
orthogonal box = (0 0 0) to (49.9998 49.9998 49.9998)
1 by 1 by 1 MPI processor grid
reading atoms …
378 atoms
read_data CPU = 0 secs
Lattice spacing in x,y,z = 3.52 3.52 3.52
ERROR: Cannot read_data without add keyword after simulation box is defined (…/read_data.cpp:302)
Last command: read_data Ni.data
Or, if I place the nickel surface .data file first, I get the following:
LAMMPS (7 Aug 2019)
OMP_NUM_THREADS environment is not set. Defaulting to 1 thread. (…/comm.cpp:93)
using 1 OpenMP thread(s) per MPI task
OMP_NUM_THREADS environment is not set. Defaulting to 1 thread. (…/comm.cpp:93)
using 1 OpenMP thread(s) per MPI task
Lattice spacing in x,y,z = 3.52 3.52 3.52
Reading data file …
orthogonal box = (0 0 0) to (200 200 200)
1 by 1 by 1 MPI processor grid
reading atoms …
370386 atoms
read_data CPU = 0.627239 secs
ERROR: Cannot read_data without add keyword after simulation box is defined (…/read_data.cpp:302)
Last command: read_data Cu.data
When I add the “add” keyword, I get the following (abbreviated):
ERROR: Illegal read_data command (…/read_data.cpp:144)
Last command: read_data Ni.data add
Now, if I modify the code to only view the nickel surface, I get:

And now, only the copper surface:
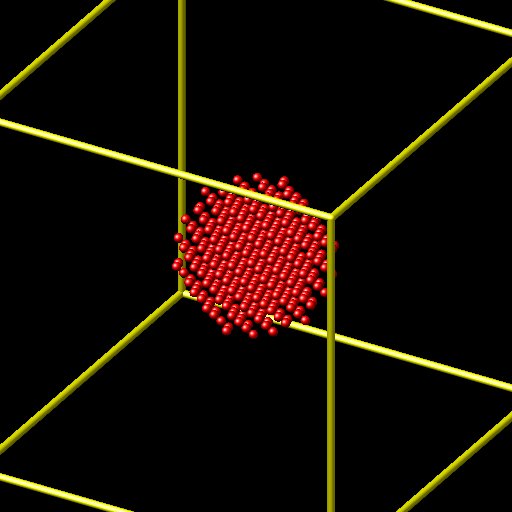
I don’t understand the error. I know that the files are correct separately, but when I combine them in the simulation, I have problems. Should I update LAMMPS perhaps? Or should it work the same?
In my understanding, the error might be in this part, but I’m not sure what exactly is wrong or how to fix it:
atom_style atomic
atom_modify map array
lattice fcc 3.52
region box block 0 40.0 0 40.0 0 40.0 units lattice # Here I’m trying to create the region #where the collision will take place
#create_box 3 box
read_data Ni.data
read_data Cu.data
Your help would be greatly appreciated. I’m stuck on this and your attention would be of great help. Thank you very much.