Dear Lammps Developer and users,
I am writing to you today to express my concern about the Non-numeric atom coords error that I am encountering when applying external electric field in reaxff.
I have been working with different electric field intensities, and the system responds to some of the efield, while gives the Non-numeric errors for others. The only difference in all the input scripts is just the efield magnitude value.
I am considering that there isn’t any problem with the geometry because I’m running the delete overlap command as well. No atoms were deleted. So the system working for few magnitudes and not having any atoms overlapping, what can be the issue ?
I am concerned that this error may be preventing me from accurately simulating the effects of external electric field on my system. I would be grateful if you could provide any suggestions on how to troubleshoot this issue.
This is my efield command in input script, I’m only changing the magnitude of z direction. Here it is 25MeV/cm. It doesn’t work. For 5, 10, 20 MeV/cm, I’m not having any issues.
fix 3 efieldapply efield 0.0 0.0 5.755 region efieldarea # conversion 0.2302*MeV/cm ## Change This ##
Isn’t reaxff using different unit for efield ?
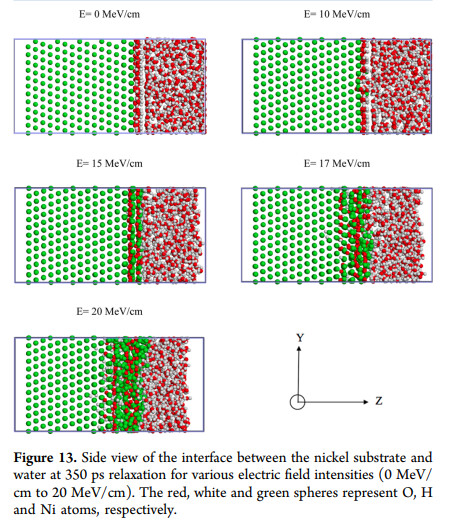
I’m currently replicating the simulations of H. DorMohammadi et al. / Computational Materials Science 145 (2018) 126–133 in “Atomistic simulation of initial stages of iron corrosion in pure water
using reactive molecular dynamics”.
Can you help with this ?
That’s a (very poorly) written comment about how the code works internally (I should know, because I worked on this very function recently), and not about how the end-user is meant to interact with the code.
The documentation for fix efield clearly states to supply the electric field corresponding to the units setting, which is volts per angstrom for both real and metal. LAMMPS does many unit conversions on the back end to make that work.
Thank you for the clarity. So for confirming, if I want to create a effect of 25MeV/cm, what should be my input magnitude in V/angstrom ? Since former is energy and the later is charge.
The attached image is from the paper that I’m using as a source.
Again, MeV/cm is a unit of force, not electric field. You will have to find out whether the authors actually meant MV/cm, and you should be able to replicate their results qualitatively (for example simulating 10, 15, and 20 MV/cm, maybe using fewer Ni layers and a smaller simulation box to speed up your simulations).
Straightforwardly, 1 MV/cm = 1e8 V/m = 1e-2 V/angstrom. You should double-check this calculation for yourself.