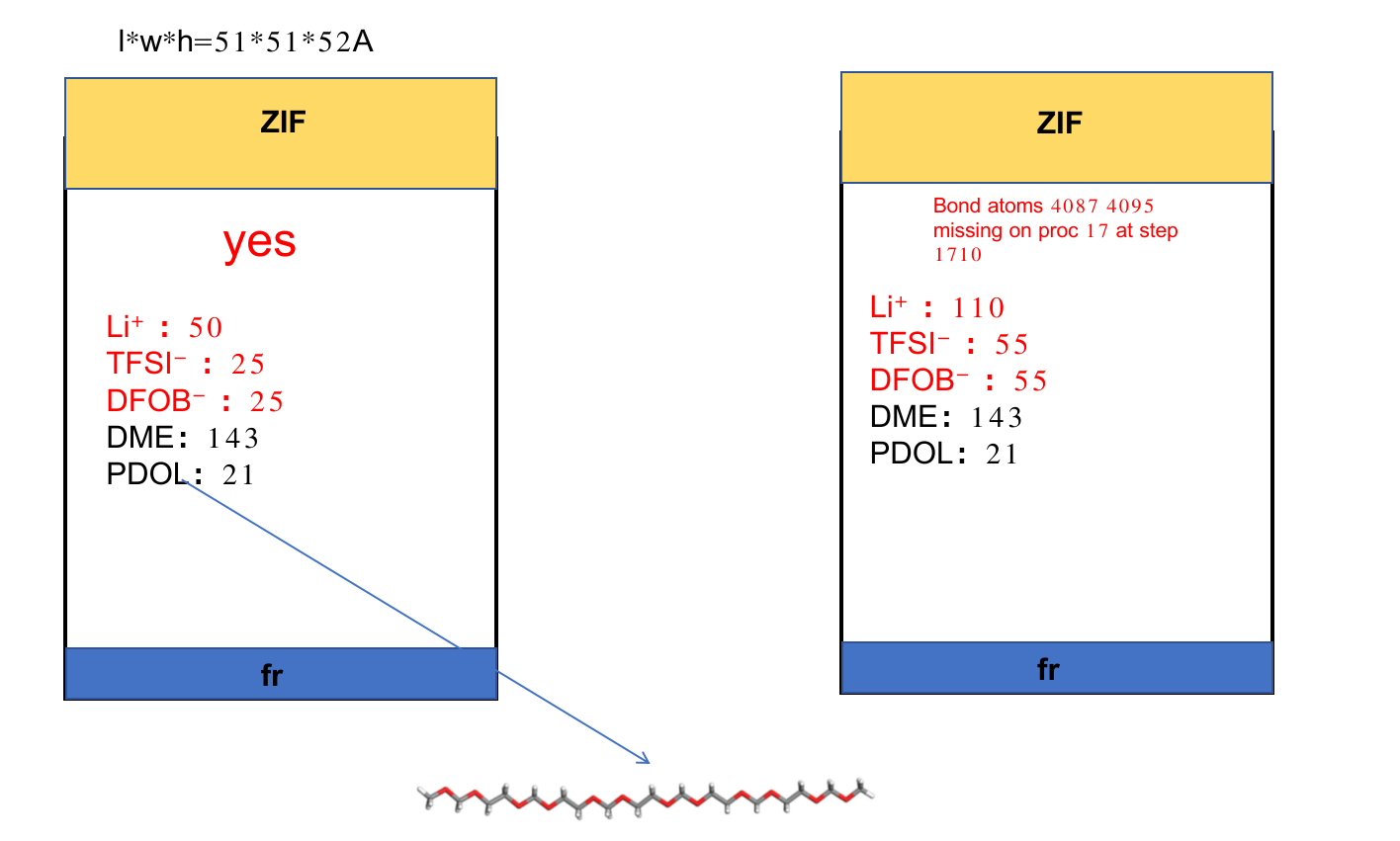
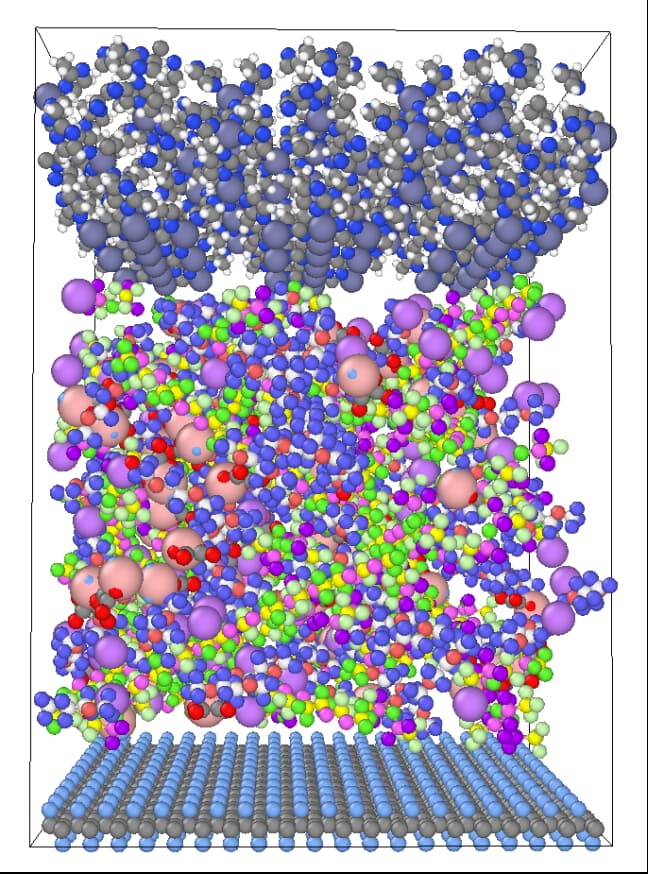
I am running the molecular dynamics of a battery electrolyte, as long as I put too many molecules in the box will report an error, and this amount is far from my target density of 1.23, I would like to humbly ask why?
In fact, I’ve tried this so many times that whenever I get too many molecules, I get an error.
Have you ever played the game Tetris?
If yes, you should know that it is quite difficult to place shaped objects into a volume so that they fill the volume densely, especially, if you place them with random orientation.
The same happens here. It will be much easier to first assemble a system at lower density and then use fix deform to slowly compress your system to the desired density. Please note that you also have to deal with physical condition called “jamming” that will make it difficult to reach the desired state, especially when you apply only molecular dynamics. You will certainly have to create a significant number of different systems and ideally use different initial conditions and different settings to find the best way to reach something approximating equilibrium.
Instead of packmol, you may want to look at the EMC software to setup your system. It was written by person that works on polymers.
2 Likes
Thank you very much for your answer, Dr. Kohlmeyer, I have solved the error.
I succeeded in making the initial model more reasonable by tweaking the packmol command. But the method you offer is much more reasonable and universal.
thank you again!!