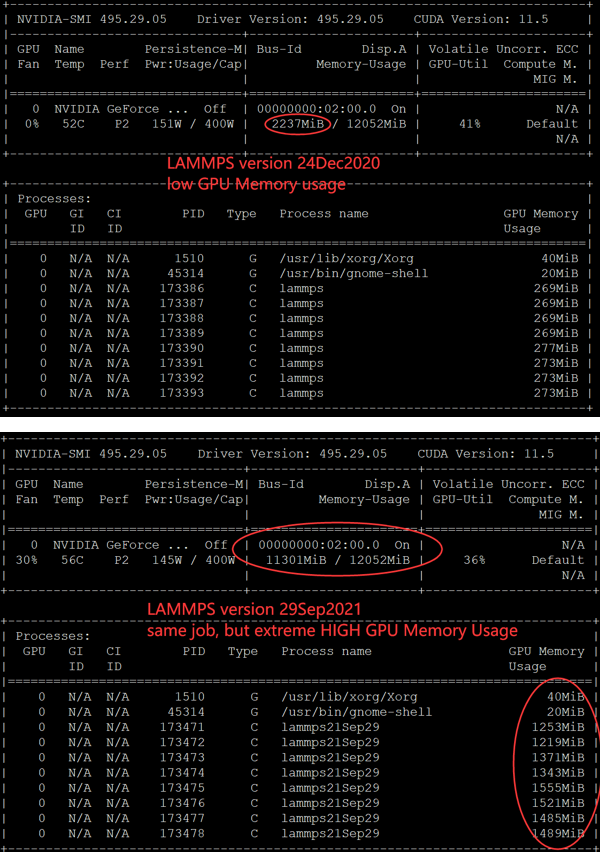
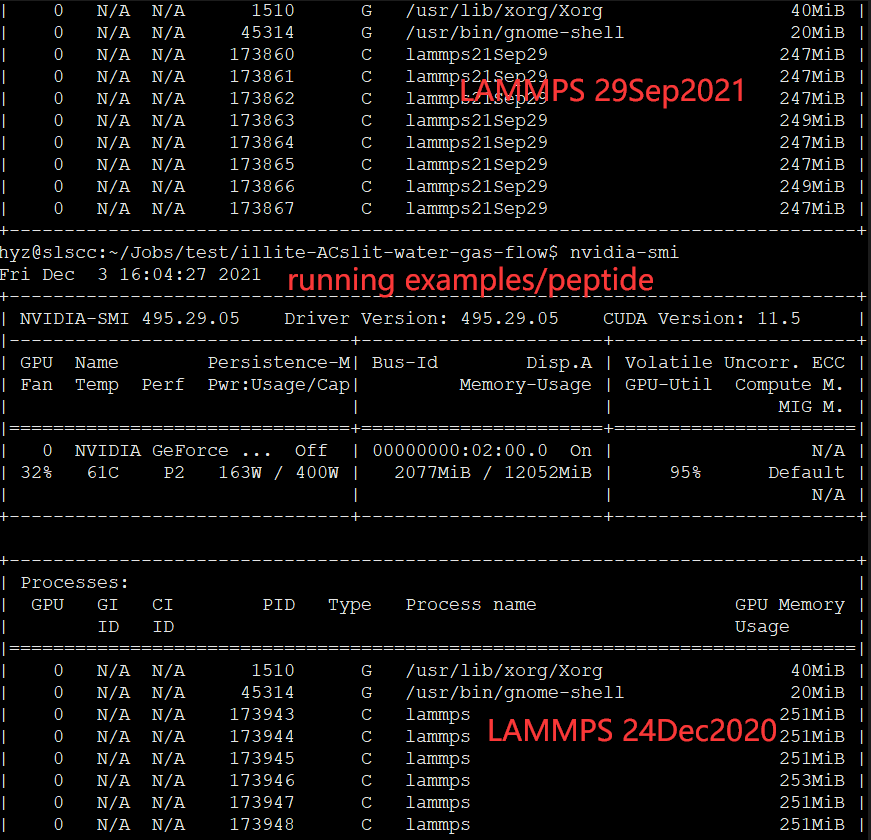
Recenly, I installed lammps-stable-29Sep2021 on Ubuntu 20.04.3 LTS (GNU/Linux 5.11.0-41-generic x86_64) with “NVIDIA GeForce RTX 3080 Ti”. However, when I run lammps job with GPU package enabled, I got an error stated that “ERROR on proc 3: Insufficient memory on accelerator”.
Belowing is the information for my lammps and systems:
(1) The lammps are installed with package GPU,OPENMP,and cuda, etc. using the CMake commands:
hyz@slscc:~/Programs/lammps-29Sep2021/build$ cmake …/cmake/ -D LAMMPS_MEM
ALIGN=64 -D FFT=FFTW3 -D PKG_COMPRESS=yes -D PKG_GPU=yes -D GPU_API=cuda -D GPU_PREC=mixed -D GPU_ARCH=sm_86 -D PKG_OPENMP=yes -D PKG_KSPACE=yes -D PKG_MOLECULE=yes -D PKG_RIGID=yes -D WITH_GZIP=yes -D WITH_JPEG=yes -D WITH_PNG=yes -D WITH_FFMPEG=yes -D FFMPEG_EXECUTABLE=/usr/local/bin
hyz@slscc:~/Programs/lammps-29Sep2021/build$ cmake --build ./ -j 32
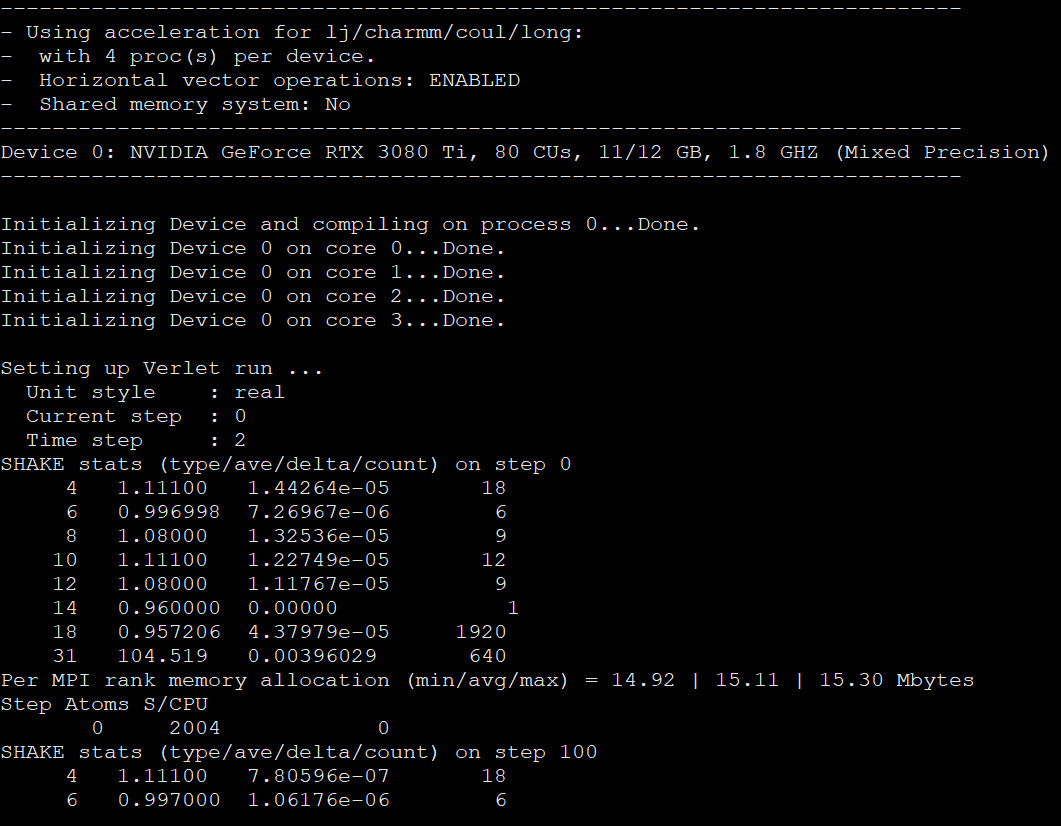
the lammps building process is ok as shown in the picture below:
when I type “lammps -help”, detailed lammps exe infor is shown below:
OS: Linux “Ubuntu 20.04.3 LTS” 5.11.0-41-generic on x86_64
Compiler: GNU C++ 9.3.0 with OpenMP 4.5
C++ standard: C++11
MPI v3.1: Open MPI v4.1.2, package: Open MPI hyz@slscc Distribution, ident: 4.1.2, repo rev: v4.1.2, Nov 24, 2021
Accelerator configuration:
GPU package API: CUDA
GPU package precision: mixed
OPENMP package API: OpenMP
OPENMP package precision: double
Compatible GPU present: yes
Active compile time flags:
-DLAMMPS_GZIP
-DLAMMPS_PNG
-DLAMMPS_JPEG
-DLAMMPS_FFMPEG
-DLAMMPS_SMALLBIG
sizeof(smallint): 32-bit
sizeof(imageint): 32-bit
sizeof(tagint): 32-bit
sizeof(bigint): 64-bit
Installed packages:
COMPRESS GPU KSPACE MOLECULE OPENMP RIGID
(2) the linux system is Ubuntu 20.04.3 LTS (GNU/Linux 5.11.0-41-generic x86_64)
(3)The GPU cuda is installed from the Nvidia web site following its instructions. The cuda version is 11.5 along with the driver 495.44 as shown below:
*hyz@slscc:~$ nvidia-smi *
*Thu Dec 2 16:42:34 2021 *
±----------------------------------------------------------------------------+
| NVIDIA-SMI 495.44 Driver Version: 495.44 CUDA Version: 11.5 |
|-------------------------------±---------------------±---------------------+
| GPU Name Persistence-M| Bus-Id Disp.A | Volatile Uncorr. ECC |
| Fan Temp Perf Pwr:Usage/Cap| Memory-Usage | GPU-Util Compute M. |
| | | MIG M. |
|===============================+======================+======================|
| 0 NVIDIA GeForce … Off | 00000000:02:00.0 On | N/A |
| 0% 38C P8 28W / 400W | 63MiB / 12052MiB | 0% Default |
| | | N/A |
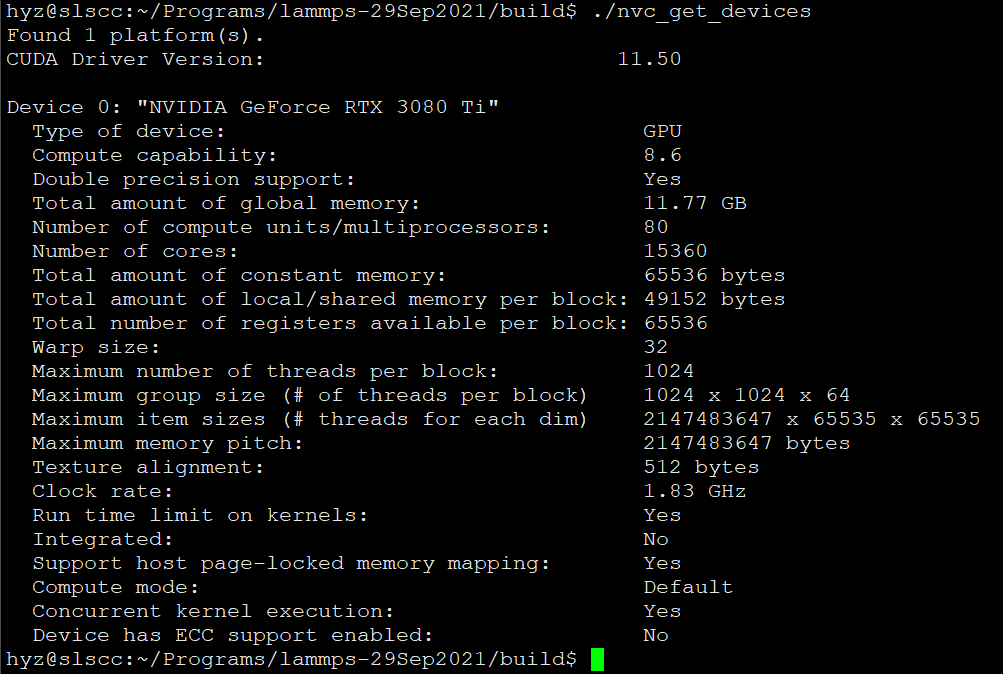
and the cmd “./nvt_get_devices” showed this as in the picture:
(4) Jobs running using lammps:
I prepared the lammps input script that contained:
if $${pk} ==gpu then &
package gpu 1 neigh no newton off split 1.0" &
elif $${pk}==cpu package omp 4" (Note: $$ is one dollar sign in this web post)
Then I type the cmd-line with either GPU or CPU (omp):
test1: $ mpirun -np 4 lammps -in inputlammps.txt -var pk cpu
The running is OK, steps going on.
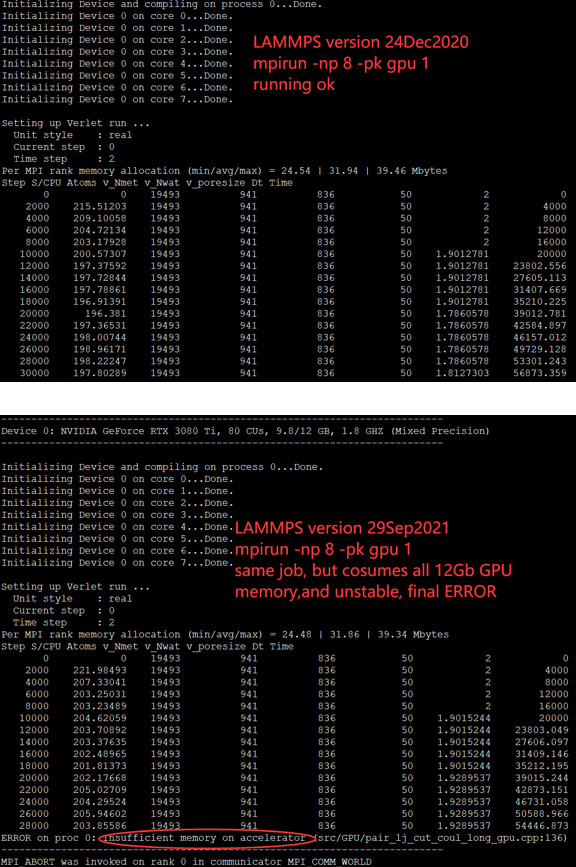
test2: $mpirun -np 4 lammps -in inputlammps.txt -var pk gpu
Then I got the error as shown in the picture below:
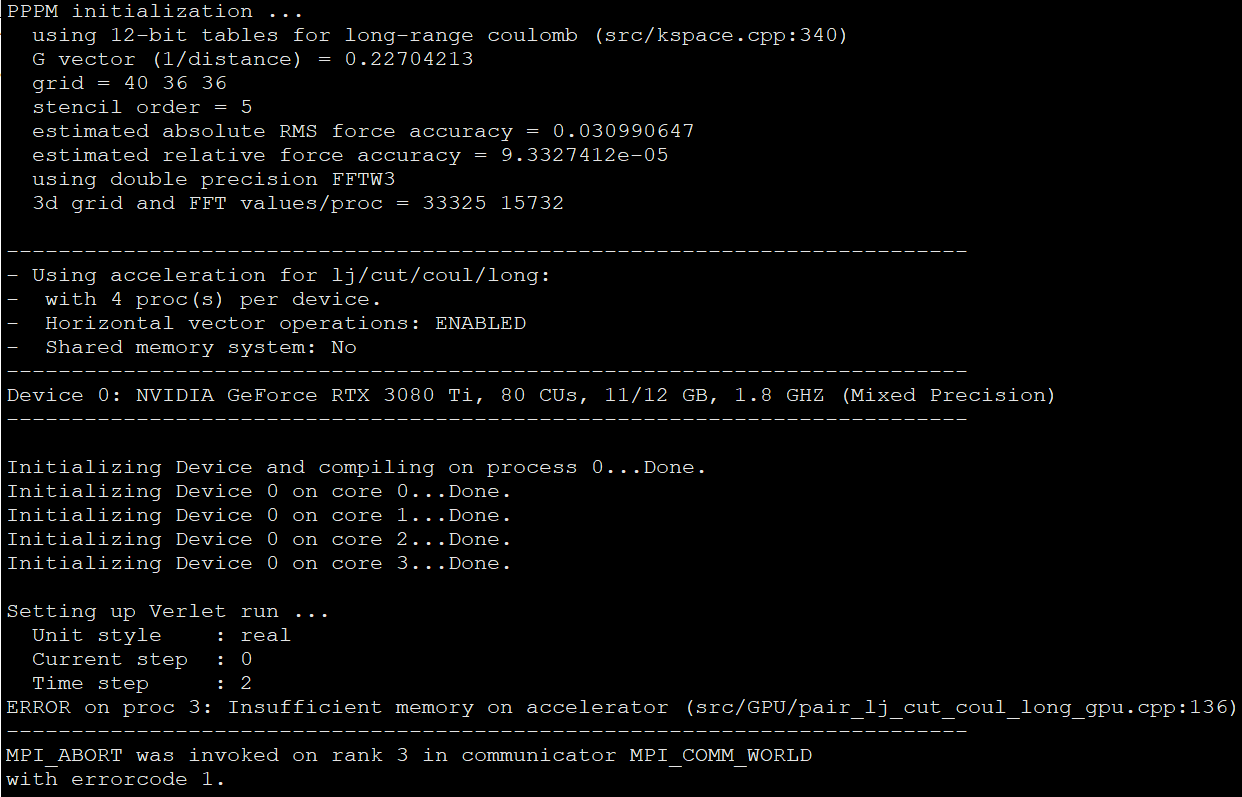
The ways I have tried are:
1>Tried many changes in the keywords of “package gpu 1 neigh no newton off split 1.0” &
2>Reinstalling the lammps with cuda_arch=sm_80 or sm_86
3>Build the lammps with traditional make
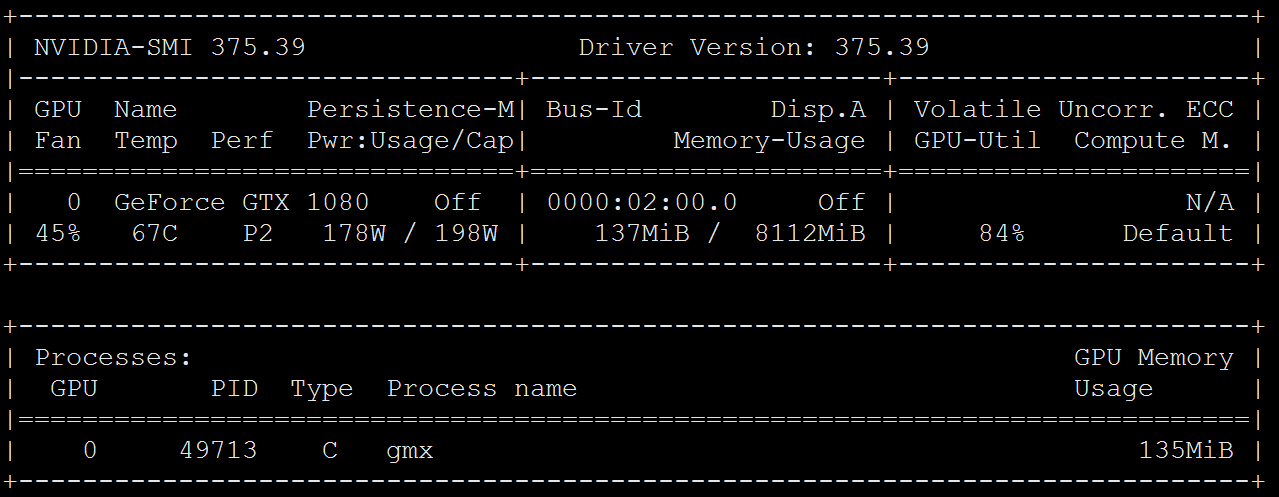
4>Additionally, the lammps with GPU cuda and mpirun went very well with the Nvidia GTX1080 on Ubuntu 16.04, which this system is now disk-wiped and reinstalled to be ubuntu 20.04 and graphics card upgraded to Nvidia GTX 3080 Ti.
5>I did some research on “shared memory” and type "ipcs -m ", it shows this:
hyz@slscc:~ ipcs -m
Shared Memory Segments
key shmid owner perms bytes nattch status
*0x00000000 18 shenliu 600 524288 2 dest *
*0x00000000 19 shenliu 600 4194304 2 dest *
*0x00000000 38 shenliu 600 4194304 2 dest *
0x00000000 39 shenliu 600 524288 2 dest
hyz@slscc:~$ free -t -m
total used free shared buff/cache available
Mem: 64190 1331 58332 5 4527 62146
Swap: 7818 0 7818
Total: 72009 1331 66151
The system have 5 users, but Shared Memory Segments showed only one user, looks weird, but I figure out nothing.
Until now, I’m struggling with this GPU error “ERROR on proc 3: Insufficient memory on accelerator”. Please indicate the way, thank you!