Hello, I am literally very new to lammps and Molecular Dynamics in general. My past works revolves within the Density functional theory framework which I use is Quantum Espresso for simulation. That said, I picked up an interest in doing molecular modelling and learning lammps for the past few days.
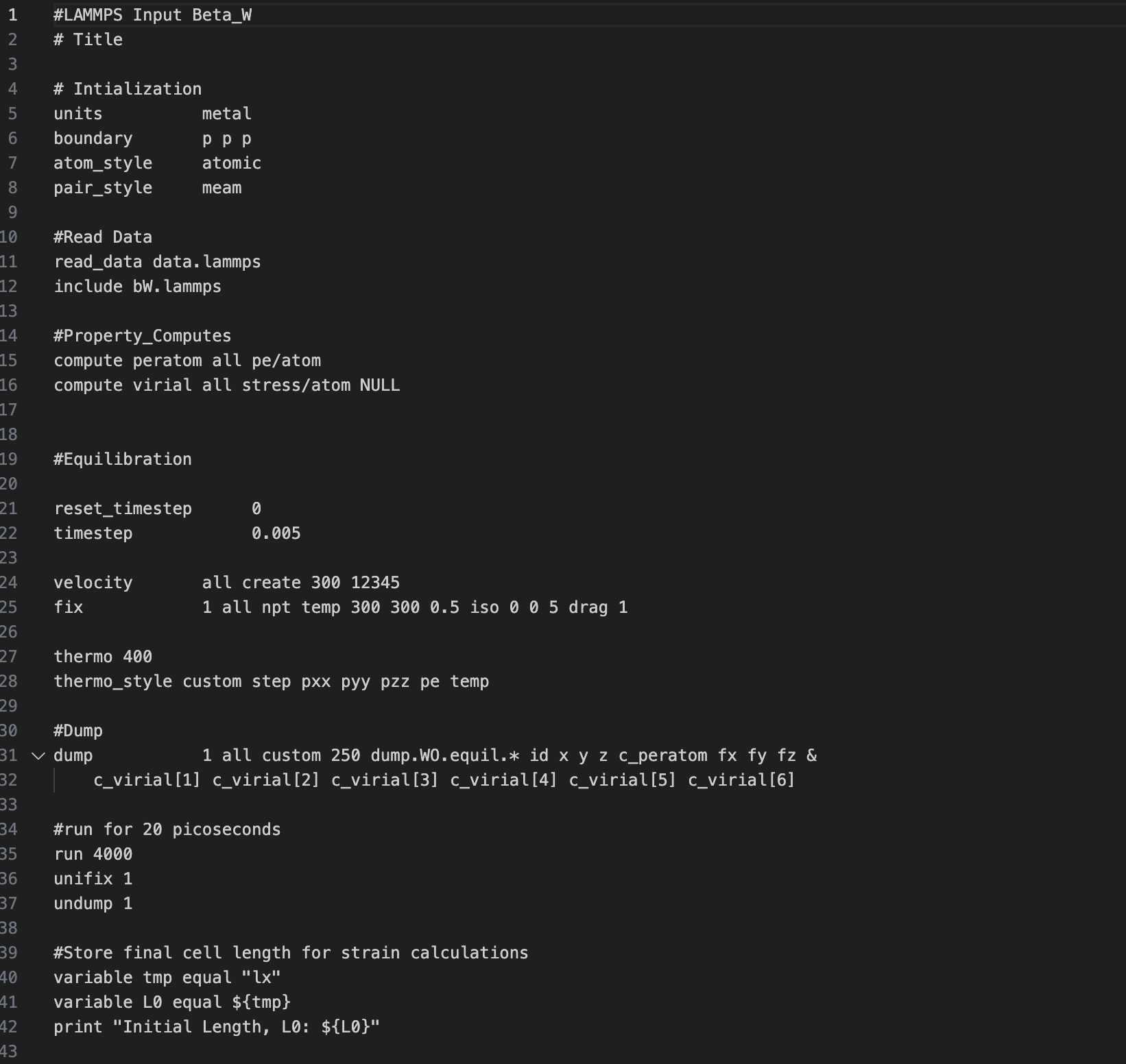
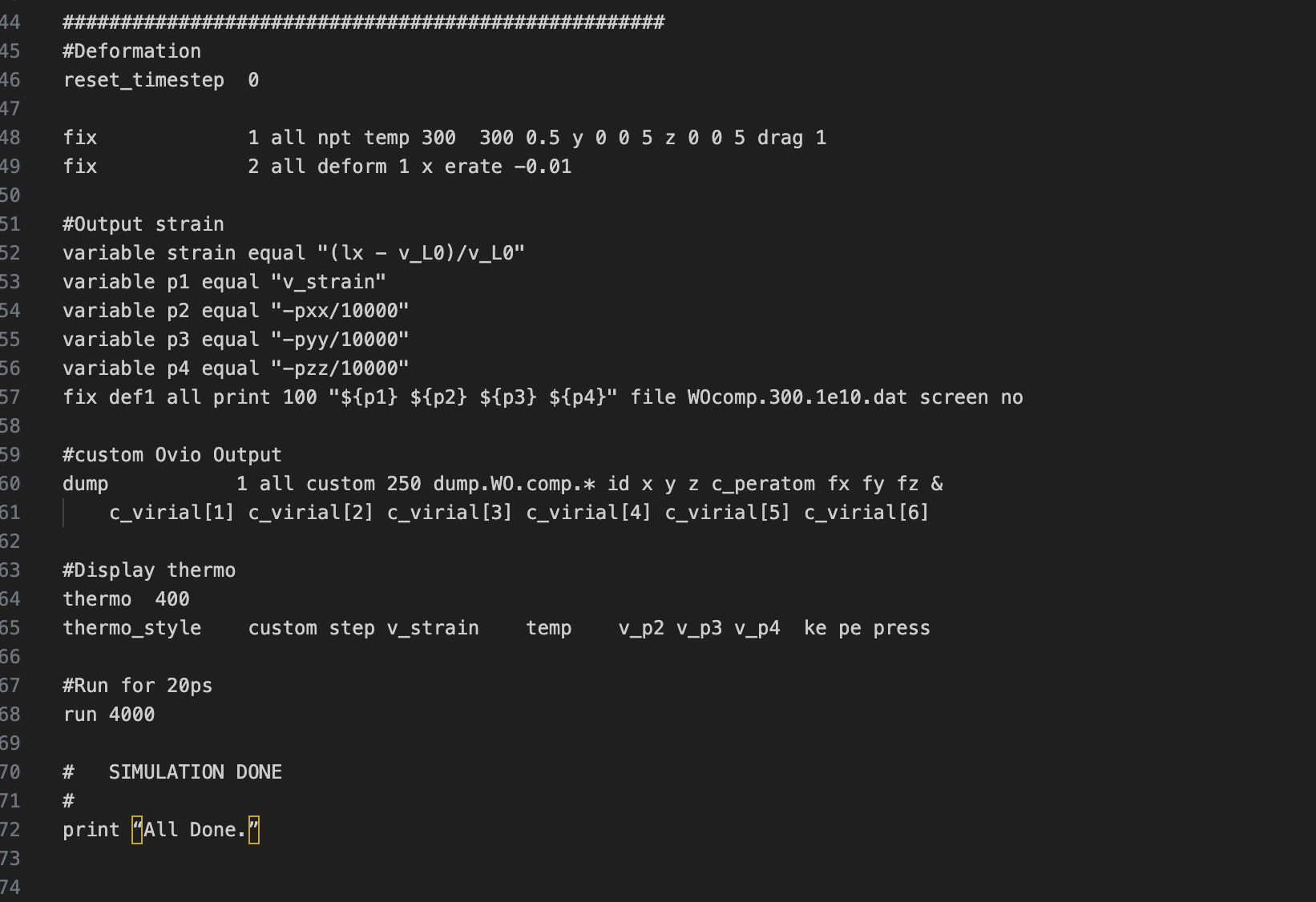
I managed to create an INPUT file for lammps based on the system I am working on. Right now, I am only using 8 atoms for my unit cell, made up of 7 tungsten atoms and 1 oxygen. Since I have DFT results, structures and data for this material I am trying to replicate via molecular dynamics and perform energy comparison.
Can you spot help spot errors and suggest corrections to make this work? I also read the commands at the lammps website unfortunately, there are still things that left me confused even after reading.
I’ll be atttaching my code below.
dIKzn5PBFuTARp7KFwGlDUGd0.png)