Hello Mr. Kohlmeyer,
As per your suggestion i tried to run the complete script with single
command at a time with several changes:
A) run until EQUILIBRATION PART
1) keeping 0.3 bin+ fix nve +fix nvt ---> output until Equilibration no
error or warning.
2) keeping 0.3 bin+fix nve ---> output until Equilibration no
error or warning
B) run complete til DEFORMATION PART
# dumping standard atom trajectories
dump 1 all atom 100 deform.lammpstrj
# dumping standard atom trajectories
dump 2 all dcd 50 deform.dcd
# 20 ps MD simulation (assuming 2fs time step)
run 20000
# SIMULATION DONE
print " All done!"
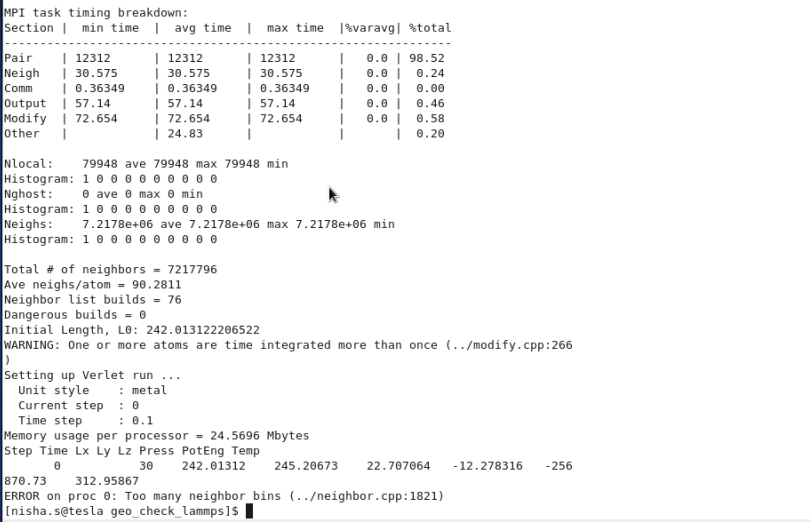
3) keeping 0.3 bin + fix nve + fix nvt ---> output WARNING: one or more
atom time integrated more than once
ERROR : too many neighbour bins
4) keeping 0.3 bin +fix nve ---> output No WARNING
ERROR : too many neighbour bins
5) keeping 2.0 nsq + fix nve ---> output No WARNING
ERROR
: too many atom sorting bins
I can not understand the problem here.
i already told you. most likely, your system is exploding for some
reason, i.e. atoms moving away extremely fast.
have you visualized your trajectory?
Please can you suggest me something on this.
you only removed one of the obvious issues, so you need to keep
removing lines from your input in a systematic fashion in order to
identify what is causing this and then it is going to be much easier
to understand why.
at any rate, this is moving into an area that you should work on with
your adviser or whoever your adviser has designated to train you. i
don't have time to do this and the lammps-users mailing list is not a
replacement to be properly trained in doing computational research.
axel.