Hi guys,
I build a polymer with pcff force filed via EMC, but the response infromation shows the total charge of the system does not zero. How can I deal this problem?
Non-zero charges can happen during typing and are typically occurring for exotic chemistries. Could you provide your .esh for which the described issue occurs?
Hi veld,
Thanks for your response to my question. Please see my .esh settings as following. Hoping for your help!
ITEM OPTIONS
replace true
field pcff
density 0.1
ntotal 41
field_increment ignore
#field_debug reduced
ITEM END
# Groups
ITEM GROUPS
pmda_oda *N1C(=O)c2cc3C(=O)N(C(=O)c3(cc2(C1(=O))))c4ccc(cc4)Oc5ccc(cc5)*,2,pmda_oda:1
caping_h *H,1,pmda_oda:1,1,pmda_oda:2
ITEM END
# Clusters
ITEM CLUSTERS
poly alternate 1
ITEM END
# Polymers
ITEM POLYMERS
poly
1 pmda_oda,1,caping_h,2
ITEM END
The error you are experiencing has to do with a quirk of EMC. EMC types per group and iterates over connections. This generally works, but seems to be problematic in certain cases, e.g. your particular case. You can circumvent the issue by creating two terminal groups, which are very similar to your original repeat unit. The latter is reflected by the following example script:
ITEM OPTIONS
replace true
field pcff
field_increment warn
number true
focus true
ntotal 10
density 0.1
emc_execute true
ITEM END
ITEM GROUPS
mono *N1C(=O)c2cc3C(=O)N(C(=O)c3(cc2(C1(=O))))c4ccc(cc4)Oc5ccc(cc5)*,1,mono:2
term1 N1C(=O)c2cc3C(=O)N(C(=O)c3(cc2(C1(=O))))c4ccc(cc4)Oc5ccc(cc5)*,1,mono:1
term2 *N1C(=O)c2cc3C(=O)N(C(=O)c3(cc2(C1(=O))))c4ccc(cc4)Oc5ccc(cc5),1,mono:2
ITEM END
ITEM CLUSTERS
poly alternate,1
ITEM END
ITEM POLYMERS
poly
1 mono,1,term1,1,term2,1
ITEM END
Dear Pieter
Thank you for debuging this problem for me. it works under the pcff force field.
However, when I replace the pcff with opls, the simlar error occurs.

Dear user,
The observed error can indicate missing rules. In EMC OPLS force fields differs from force fields as PCFF and CHARMM, such that partial charges are directly assigned through typing rules and not by means of bond increments. The error you are experiencing typically results from missing typing rules for your particular case. This means, that you will have to determine which rules are missing. You can force EMC to write out a non-neutral system by setting
system_charge false
under ITEM OPTIONS. Either the produced .emc or .psf can help you in figuring out the incorrect type and/or charge designations. Consequently, you will have to add the missing rules with the correct partial charges. It could well be, that what you are looking for is hidden in the OPLS parameter source files in ./field/opls/2012/src. You could add rules to opls.define and run this file with emc_opls.pl to obtain new .prm and .top files. Better instead would be to transform the OPLS force field into a field using bond increments for partial charges. That, however, will be a future project.
Dear Pieter
Once again, thanks for your response, and I will try it according to your tips.
Sincerely,
LIU
@veld
Hi Veld,
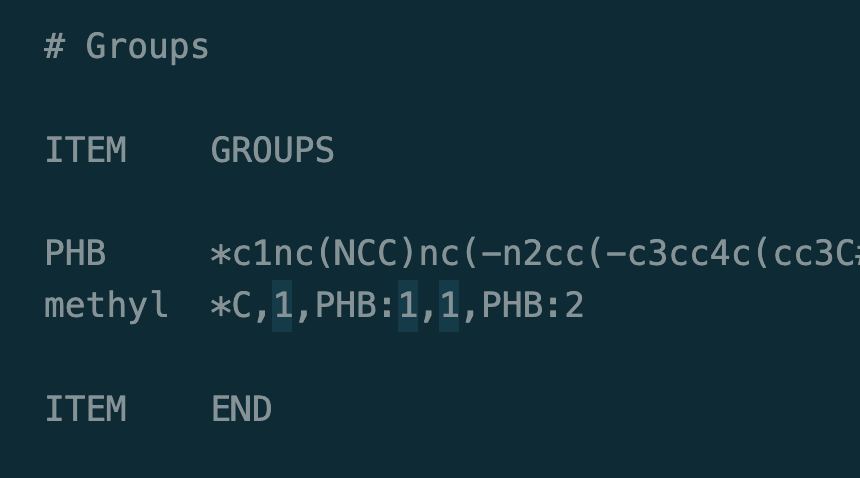
I am running into the same issue but my monomer doesn’t look as clean to implement the solution you detail above. How do you suggest i go about circumventing this issue with this monomer?
smiles:
*c1nc(NCC)nc(-n2cc(-c3cc4c(cc3C#N)Oc3cc5c(cc3O4)C3(C)c4ccc(C)cc4C5(C)c4cc5c(cc43)Oc3cc(-c4cn(*)nn4)c(C#N)cc3O5)nn2)n1
Hi Brandon,
What force field are you using? OPLS or PCFF?
Im using PCFF!