Hi,
I was trying to compile the example problem provided in lammps/examples/COUPLE/simple/simple.cpp .
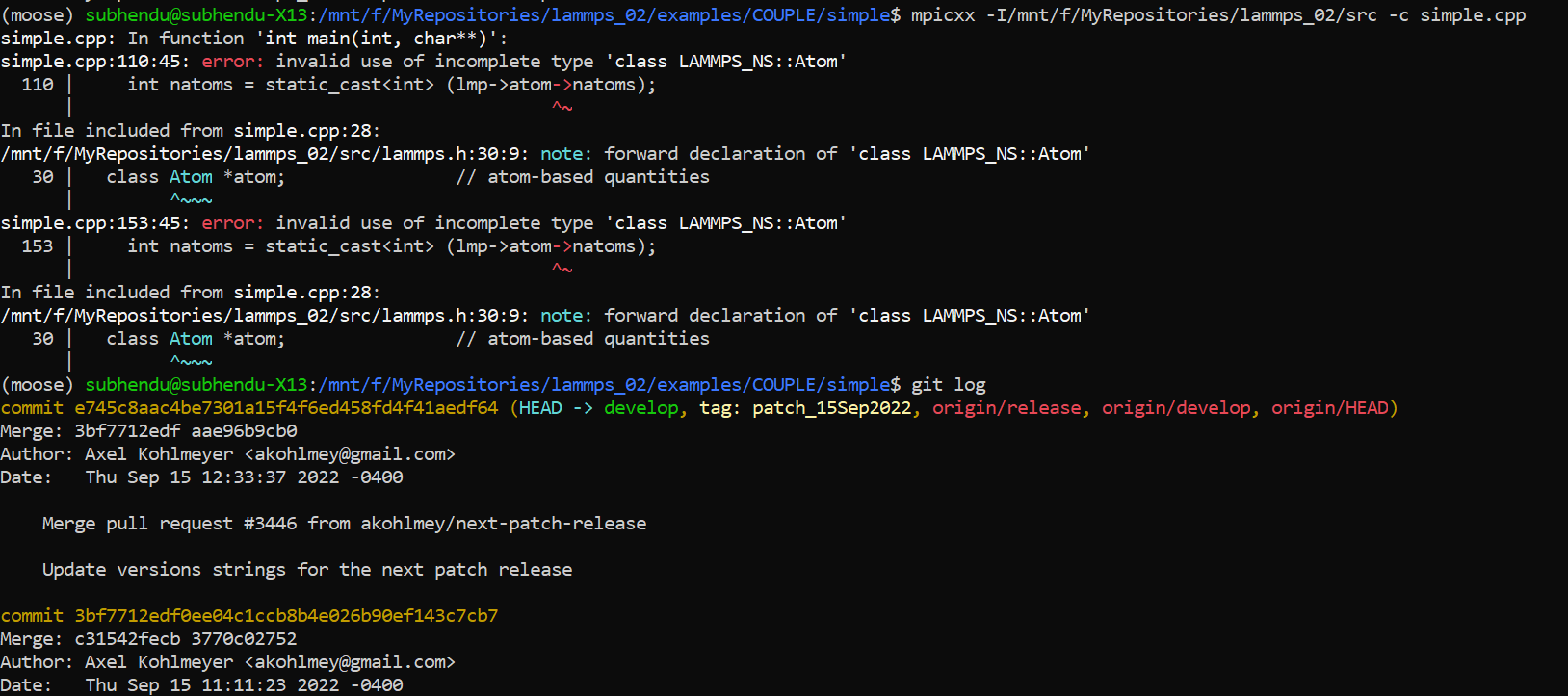
Before compiling, I created the static library of LAMMPS (liblammps_mpi.a). But even before linking that library, while compiling the ‘simple.cpp’ file I am getting this error: "simple.cpp:110:42: error: invalid use of incomplete type ‘class LAMMPS_NS::Atom’ "
The error is in line “int natoms = static_cast (lmp->atom->natoms);” at the second dereference operator (image attached).
I am using the following command for compilation (as mentioned in the ‘README’ in that folder):
mpicxx -I/mnt/f/MyRepositories/LAMMPS_01/src/ -c simple.cpp
I am getting the same error if I want to access any other variables (e.g, nlocal, nghost) declared within the “Atom” class, even though the header file ‘atom.h’ is included in the ‘simple.cpp’ and also in ‘lammps.cpp’ where the object “atom” of type “class Atom” is created.
Any idea, what is the reason and solution?
Best
Subhendu
My guess would be that you have an older version of the LAMMPS sources. Which LAMMPS versions is this with. I cannot reproduce this with the latest LAMMPS version 15 Sep 2022.
I pulled the latest version of LAMMPS (develop branch) and tried to compile but it gives the same error.
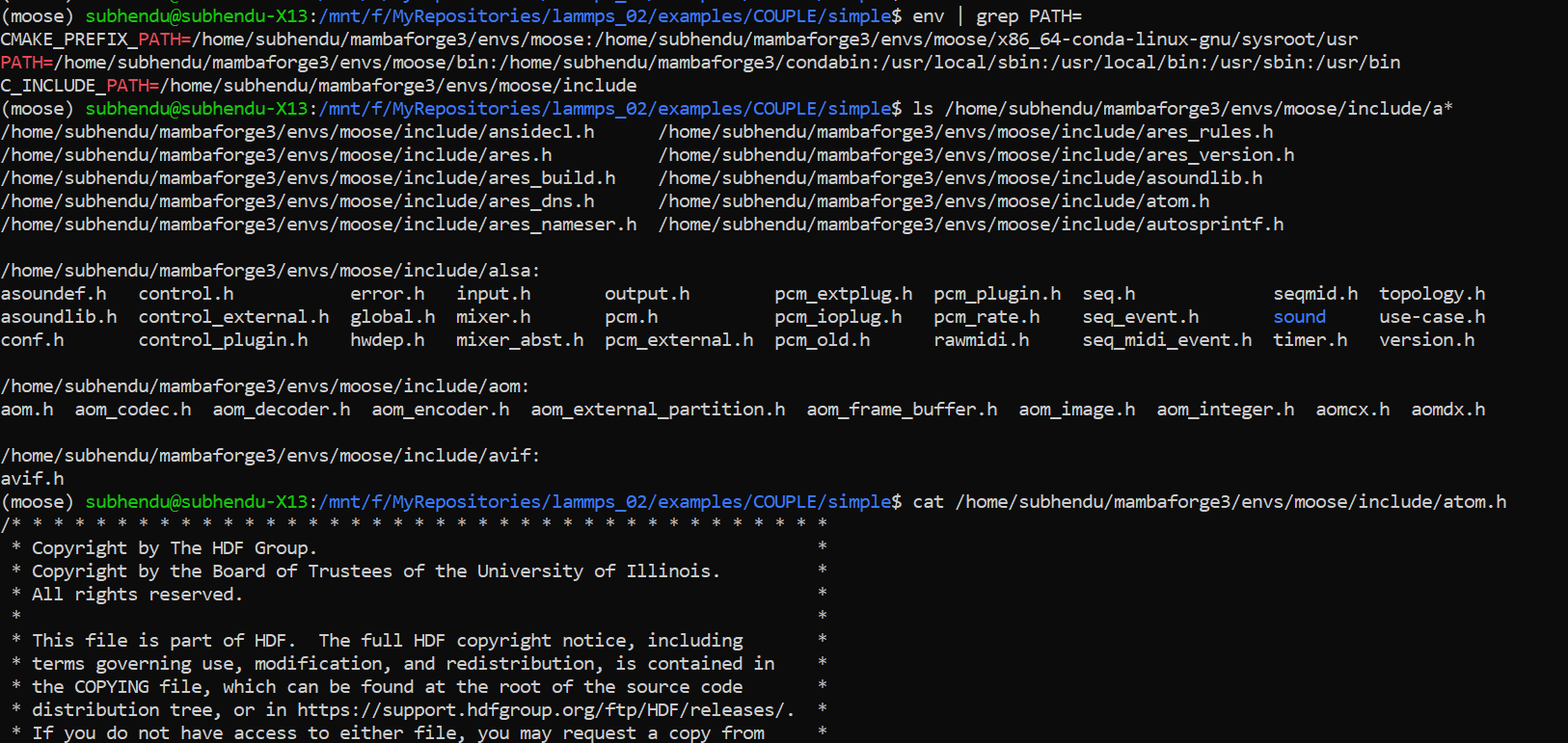
The only way I could imagine that you would get this kind of error with the commands you use, would be when you have a file atom.h from a different project/software somewhere in a folder that is included before the LAMMPS one. What is the output of
env | grep PATH=
Another test you could make would be to modify the src/atom.h file to add the #warning line as follows:
#ifndef LMP_ATOM_H
#define LMP_ATOM_H
#include "pointers.h"
#warning "Loading LAMMPS version of atom.h"
#include <map>
#include <set>
If the correct atom.h file is picked up, you will get a warning like this:
In file included from simple.cpp:30:
/home/akohlmey/compile/lammps/src/atom.h:19:2: warning: #warning Loading LAMMPS version of atom.h [-Wcpp]
19 | #warning Loading LAMMPS version of atom.h
| ^~~~~~~
If you do not get such a warning, then my hypothesis is confirmed and you have to correct your setup.
Another modification you can make to confirm the conflict of include file names would be to do the following:
- modify
simple.cpp to include atomxxx.h instead of atom.h
- in the LAMMPS source folder, add a symbolic link from
atom.h to atomxxx.h with ln -s atom.h atomxxx.h
Compiling the the example should now access the atom.h file via the symbolic link assuming that you don’t also have a duplicate/bogus atomxxx.h file somewhere.
Yes, you are right.
There was a header file with the same name ‘atom.h’ in the system search path that was causing the problem. That file was from another program placed within that conda environment. Soft linking of the lammps atom.h file helped to identify the issue.
Creating a separate conda environment with only MPI package solves the problem.
Thanks for your help.
Subhendu
LANL, Los Alamos.