Dear Community.
Could you please help me look at the problems I am currently facing?
My system is to simulate the pyrolysis of organic matter at high temperature under Reax.ff, specifically pyrolytic glucose (model is 500 glucose molecules). However, two unresolvable errors were encountered.
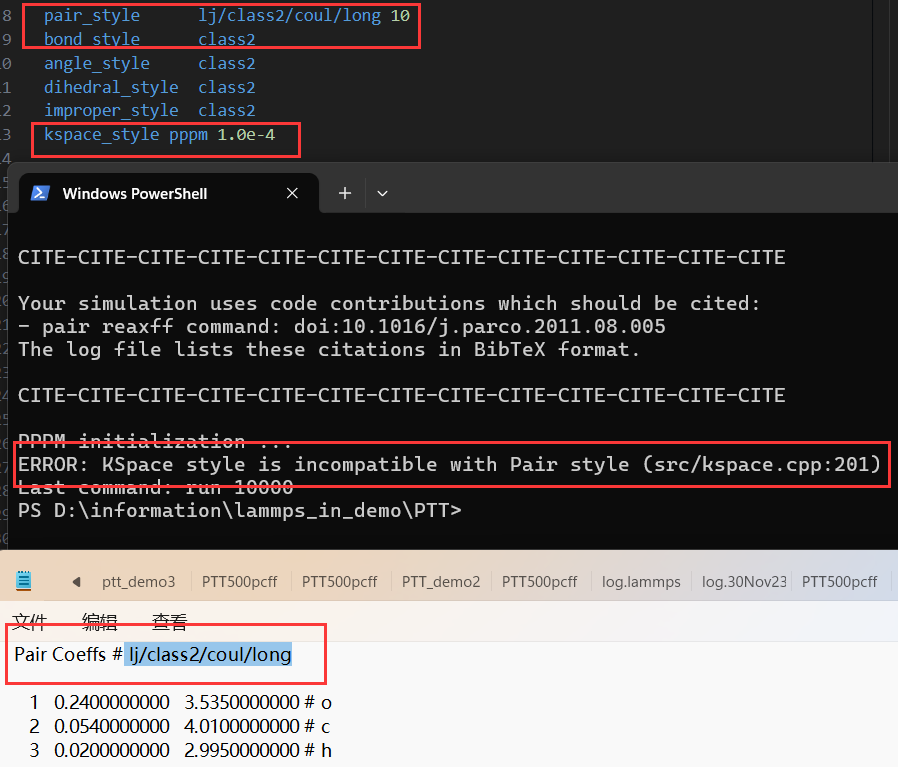
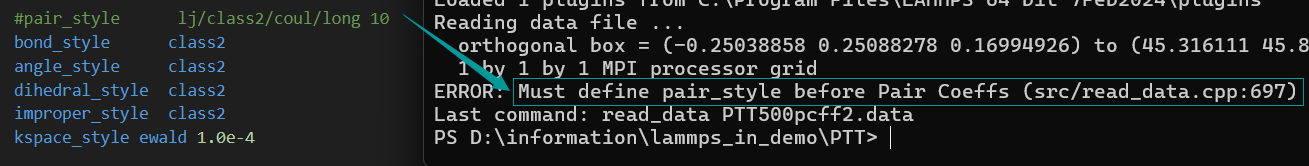
- When I run ‘kspace_style’, the error is’ KSpace style is incompatible with Pair style '.
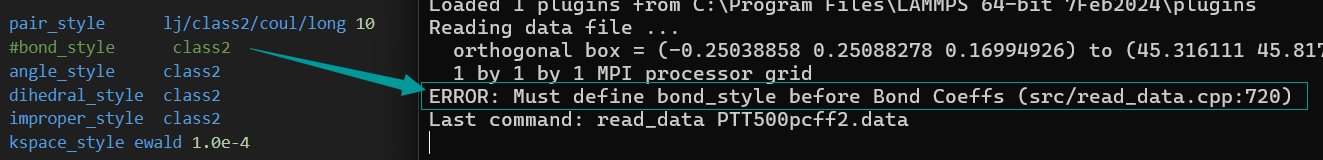
- When I annotated it, there was another error involving Bond atoms missing.
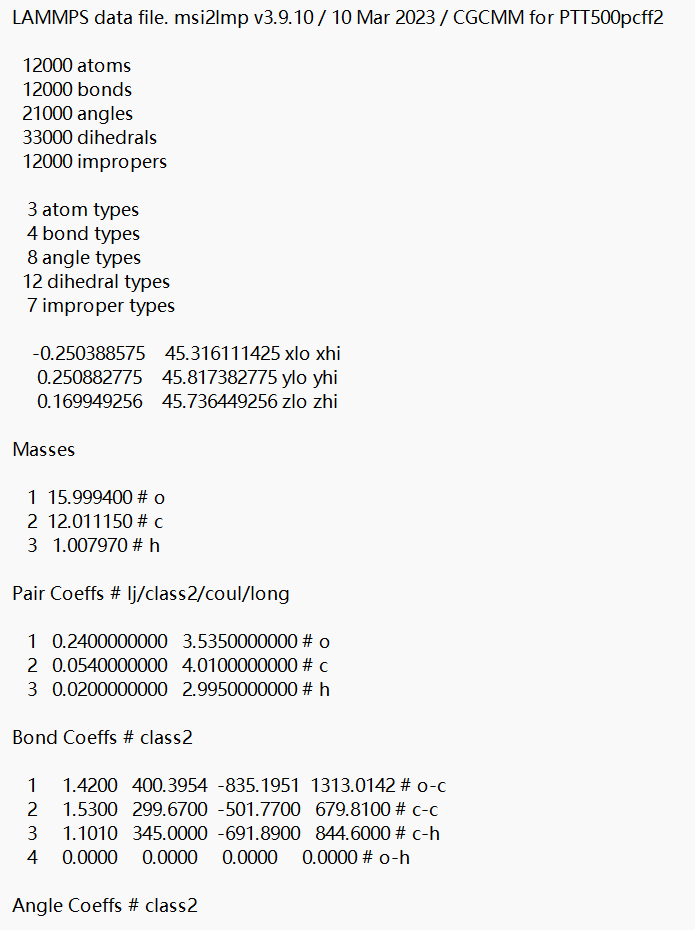
The following is my in file, I used material studios software for my modeling. I checked the paircoeff in the data file, still no solution to error.
Could you give me some suggestions on how to revise it? Thank you very much for every suggestion.
This is my first time Posting, sorry for some syntax issues and formatting issues
Best regards,
Ripples916
units real
dimension 3
boundary p p p
atom_style full
neighbor 2.0 bin
neigh_modify every 1 delay 0 check no
pair_style lj/class2/coul/long 10
bond_style class2
angle_style class2
dihedral_style class2
improper_style class2
#kspace_style pppm 1.0e-4
read_data PTT500pcff2.data
pair_style reaxff lmp_control
pair_coeff * * ffield.reax.cho H C O
timestep 0.1
velocity all create 1000.0 4928459 rot yes dist gaussian
thermo 100
thermo_style custom step temp pe ke
thermo_modify flush yes
dump 1 all custom 1000 dump-eq.dsw id type q x y z
dump_modify 1 sort id
dump 2 all xyz 1000 dump-eq.xyz
dump_modify 2 sort id
fix 1 all nve
fix 2 all temp/berendsen 1000.0 1000.0 50.0
fix 3 all qeq/reaxff 1 0.0 10.0 1.0e-6 reaxff
fix 4 all reaxff/species 1 1 1000 species-eq.txt element O C H
fix 5 all reaxff/bonds 100 bonds.reaxff
undump 1
undump 2
unfix 1
unfix 2
#unfix 3
unfix 4
unfix 5
#######################################
reset_timestep 0
timestep 0.25
dump 1 all custom 4000 dump.dsw id type x y z #
dump_modify 1 sort id
dump 2 all xyz 4000 dump.xyz
dump_modify 2 sort id
fix 1 all nve
fix 2 all temp/berendsen 2000.0 2000.0 50.0
fix 3 all qeq/reaxff 1 0.0 10.0 1.0e-6 reaxff
fix 4 all reaxff/species 1 1 1000 species-eq.txt element O C H
fix 5 all reaxff/bonds 100 bonds.reaxff
run 10000
PS:
- In order to better describe my problem, the fourth change provides information about Kspace_style, marked with a red box.
- As for the problem of bond atom missing, according to my current understanding, I only know that it is related to the atom of material studio file exceeding the border, but I do not know how to solve it specifically, so the ovito model diagram is attached.