Dear community,
I’m sorry to bother you. But after many times of debugging, I still can not solve the problem of error. So I had to ask for your help again. (Again, sorry for wasting everyone’s time.)
What I’ve done is pyrolytic PI films to simulate laser production of graphene, and the force field is ReaxFF Force field, using a version of LAMMPS 64-bit 2Aug2023-MSMPI for windows. However, an error was reported at runtime.
LAMMPS (2 Aug 2023 - Update 3)
job aborted:
[ranks] message
[0] application aborted
aborting MPI_COMM_WORLD (comm=0x44000000), error 1, comm rank 0
[1-3] terminated
---- error analysis -----
[0] on LAPTOP-K8KKF285
lmp aborted the job. abort code 1
---- error analysis -----
After checking the in file for several times, I made different concatenation between the wrong file and the file that could run successfully before, so as to find out the cause of the error.
Since most of the programs in the initialization phase were copied directly from files that worked successfully, the reason was to focus on modeling and force field Settings. The following are the details of the data file and the Force field file.
My modeling was done using Material studios software, converted into data files using msi2lmp program under pcff force field. The vmd software is then used to change the atomic type into charge. Here is the data file and its corresponding OVITO image.
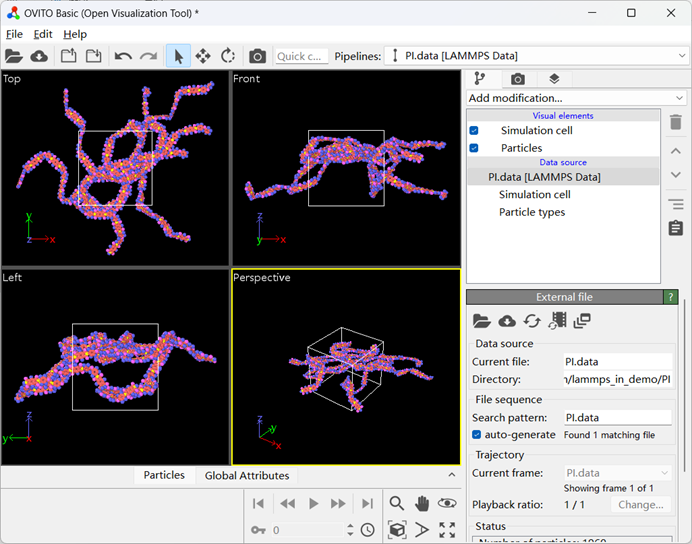
PI.data (98.0 KB)
For the force field file, my force field file is downloaded from Included force fields — ReaxFF 2023.1 documentation (scm.com), from the literature:
L.Z. Zhang, S.V. Zybin, A.C.T. van Duin, S. Dasgupta, W.A. Goddard, and E.M. Kober Carbon Cluster Formation during Thermal Decomposition of Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine and 1,3,5-Triamino-2,4,6-trinitrobenzene High Explosives from ReaxFF Reactive Molecular Dynamics Simulations Journal of Physical Chemistry A (2009) 113, 10619-10640 http://dx.doi.org/10.1021/jp901353a
I converted it from a PDF file to a txt file and then manually reformatted the force field. Finally, the format was adjusted using the fromdos applet in the Ubuntu subsystem. I attached the force field file I used below.
reaxff.chon.txt (17.4 KB)
To avoid making the stupid mistake of typing the wrong file name again, I typed the dir command on the command line and compared it with the run command to rule out the cause of the error.
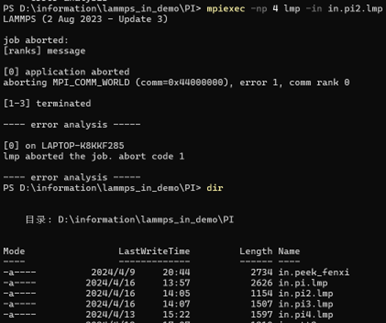
This is the second molecular dynamics model I’ve done. My study is inseparable from the support and advice of the community. Thank you very much for your advice. Any advice will benefit me a lot, and every reply will become my motivation to move forward. Sorry again for my ignorance.
Best regards,
Ripples