Dear LAMMPS users,
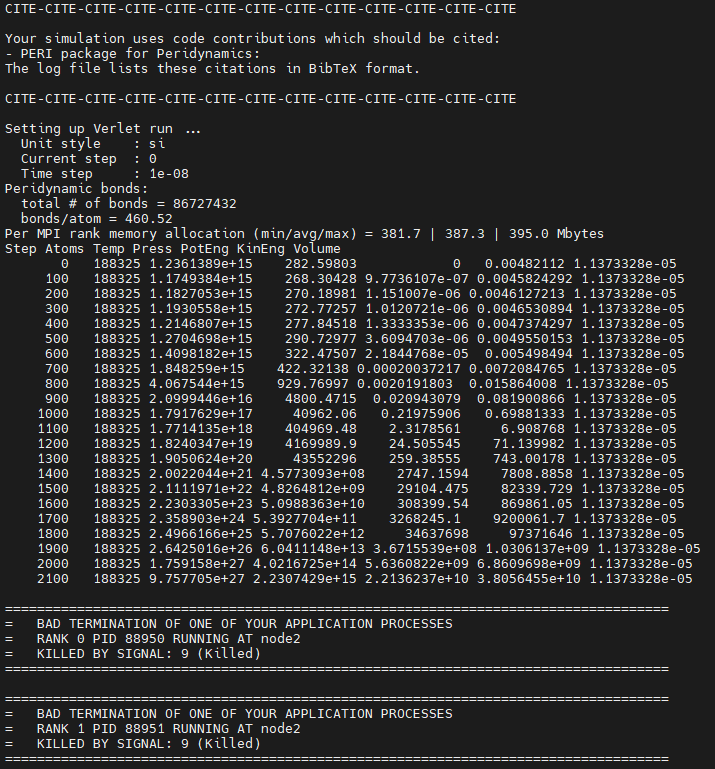
I am a novice user of the LAMMPS, currently trying to simulate the stretch of an elastoplastic system with initial crack using PERI package.However, I have encountered some errors when setting the velocity of the top several layers of particles using the “fix move” or “velocity set” command. The simulation terminated with a “killed by signal:9” error or results in “-nan” values in thermo recorded in the out.log file. (the dump output file show orderly crack between the top and other particles)
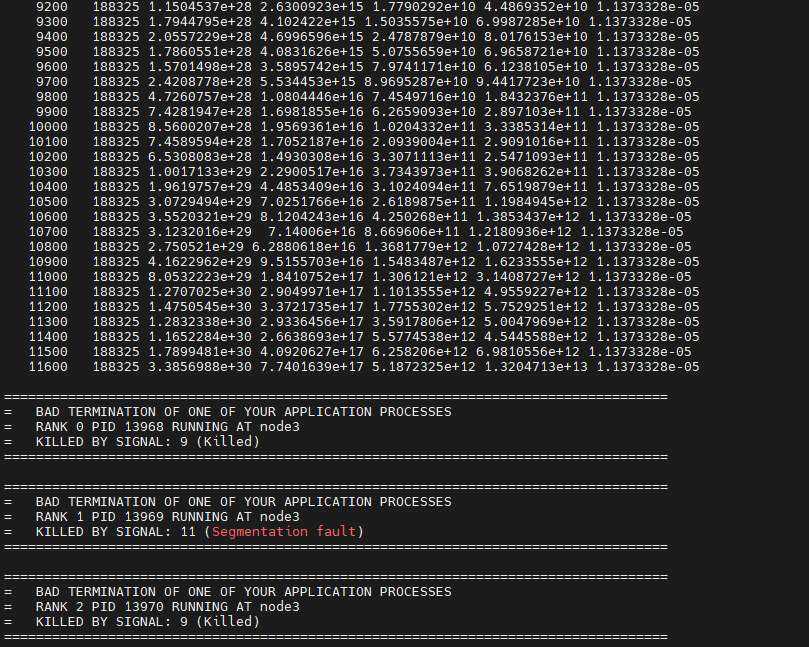
Similary, when I switched to using the “fix defoem” command and set periodic boundary conditions (p p p), a similar error “-nan” happened. (these error happened even when I tested a PMB brittle model.)
When “killed by signal:9” occurs, the simulation task is killed, and when “-nan” occurs in thermo, the simulation continues, but particles are missing from the dump output structure, leaving only the box. (error messages in attached figures)
The objective I aim to achieve is calculating crack growth for elastoplastic or hyperelastic ductile materials under stretch conditions and obtaining a stress-strain curve. Any assistance provided would be greatly appreciated.
Best Regards.
Jichun Zhao
①here is the dump file:
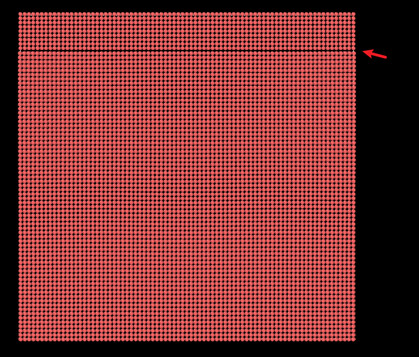
②here is the errors:
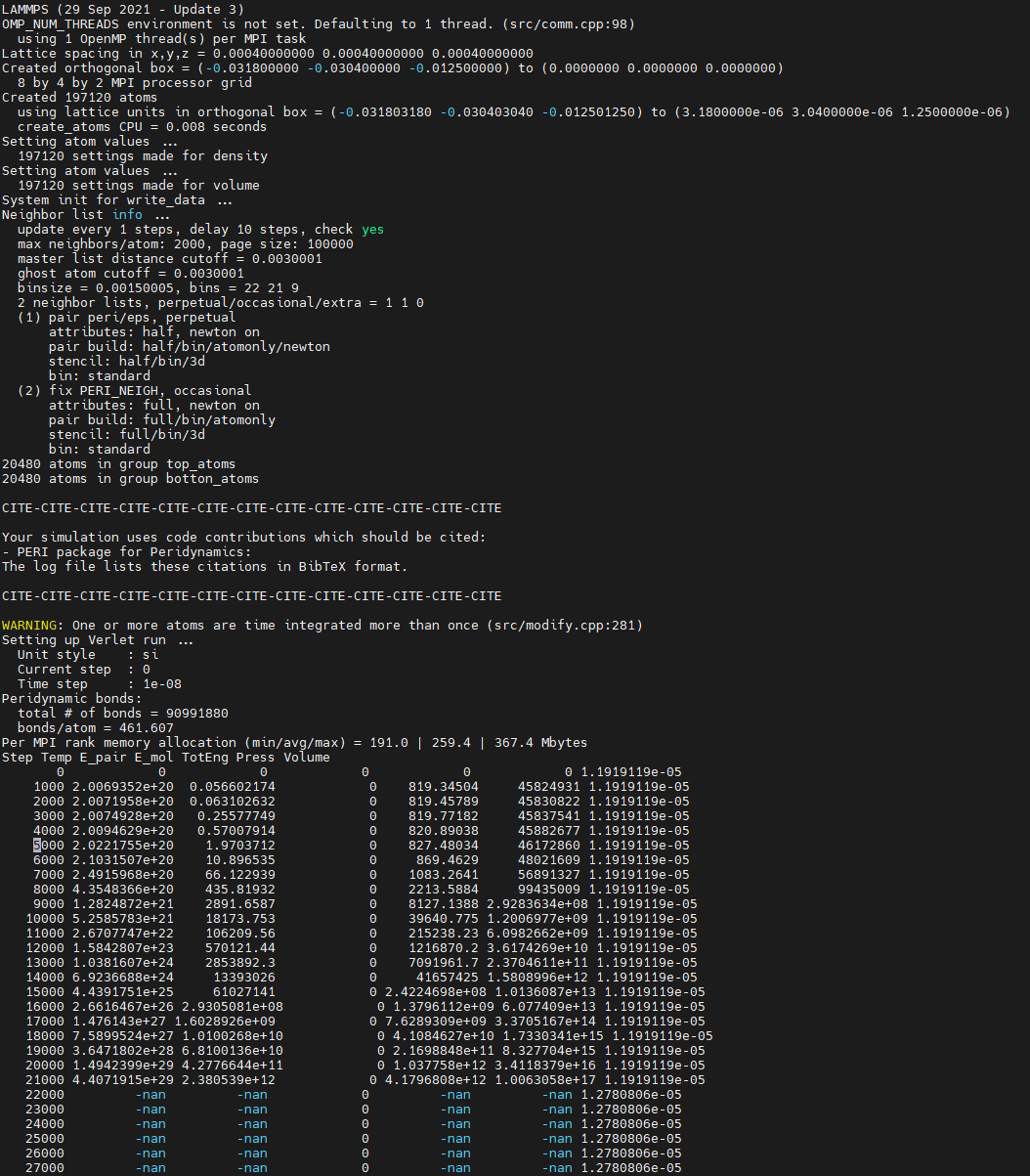
③here is the input:
3D Peridynamic simulation"
units si
dimension 3
boundary s s s
atom_style peri
atom_modify map array
neighbor 0.001 bin
#neigh_modify one 40000
lattice sc 0.0004
Create desired target 32x30x12.4mm and add atoms
region specimen block -32e-3 0.0 -30e-3 0.0 -12.4e-3 0.0 units box
create_box 5 specimen # 5-types atoms
create_atoms 1 region specimen
region boxA block -15.91e-3 0.0 -17.3e-3 -15e-3 -12.4e-3 0.0 units box
region boxB block -15.91e-3 0.0 -15e-3 -12.7e-3 -12.4e-3 0.0 units box
region boxC block -18.5e-3 -15.9e-3 -15e-3 -12.7e-3 -12.4e-3 0.0 units box
region boxD block -18.5e-3 -15.9e-3 -17.3e-3 -15e-3 -12.4e-3 0.0 units box
group a region boxA
group b region boxB
group c region boxC
group d region boxD
set group a type 2
set group b type 3
set group c type 4
set group d type 5
Peridynamic material settings for the target
pair_style peri/eps
pair_coeff * * 210E9 70e9 2.0001e-3 0.10 0.2 30e8
pair_coeff 2 3 210E6 70e6 2.0001e-3 0.01 0.2 30e8
pair_coeff 2 4 210E6 70e6 2.0001e-3 0.01 0.2 30e8
pair_coeff 3 5 210E6 70e6 2.0001e-3 0.01 0.2 30e8
Set mass density 800 kg/m^3
set group all density 800
volume = lattice constant^3 every particle volume
set group all volume 6.4e-11
Zero out velocities of particles
velocity all set 0.0 0.0 0.0 sum no units box
Use velocity-Verlet time integrator
fix F1 all nve
Compute damage for each particle
compute C1 all damage/atom
compute sigma all stress/atom NULL
compute pe_per all pe/atom # potential energy per atom
compute ke_per all ke/atom # kinetic energy per atom
write_data geo.data
stretch along y-axis
region top block -32e-3 0.0 -30e-3 0.0 -12.4e-3 0.0 units box # top boundary
group top_atoms region top
fix stretch_top top_atoms move linear 0.0 1 0.0 units box # stretch velocity
#fix stretch_box all deform 10 y final -20e-3 20e-3 units box # deform
Output total energy and kinetic energy
timestep 1.0e-7
thermo_style custom step atoms temp press pe ke vol
thermo 100
dump D1 all custom 4000 dump.peri id type x y z c_C1 c_pe_per c_ke_per #c_sigma[1] c_sigma[2] c_sigma[3] c_sigma[4] c_sigma[5] c_sigma[6]
run 20000