Hi Axel,
1. lammps-12Dec18. Running on linux, with my LAMMPS scripts using the Python interface (Python 3.6.5)
you may consider updating to a more recent LAMMPS version, although
chances are limited that what is causing your problems is addressed.
a newer version would also include more information useful for
debugging in the help message.
2. GNU g++ compiler. Running on HPC with "Each of the 525 Lenovo nx360 m5 compute nodes has two 14 core 2.4 GHz Intel E5-2680 v4 (Broadwell) CPUs, and 128 GiB of RAM."
what would be interesting to know is the interconnect hardware that
carries the communication between those nodes. that would have a
significant impact on the multi-node performance.
3. I compiled it myself but quite a long time ago so can't fully remember the process. I've attached the -help output.
4. Bus error happens after a long wall time. Running two simulations of the same system (with tiny randomised differences) will crash at a similar timestep, but not identical. All simulations seem to eventually hit this error if left long enough (i.e. if I left something to run for an infinite number of timesteps, eventually the error would occur)
this hints at some kind of memory leak or memory corruption issue
caused by migration of atoms between MPI ranks.
unfortunately, the way how you run them through the python interface
makes it *extremely* difficult to track down any issues.
this is further complicated by using a custom pair style. we do tests
on code that is included with the LAMMPS distribution to minimize the
risk of memory leaks or other related errors, but that is - of course
- impossible to external code. also we are limited by the code
coverage of the tests we have. trying to increase that systematically
(we are currently at about 25 % coverage) is an ongoing goal, but that
can take many years to get near completion and for some parts of the
code will be next to impossible.
if you can create input decks that can run without using the python
interface, you should see, if you can reproduce the bus error (or
something similar) with that input deck. that will make it easier to
debug things. then it would be imperative to determine whether this
issue is coupled to the custom pair style or can be reproduced
without. based on those findings, it would be easier to narrow down
where to look to identify and fix the memory issue.
recent versions of the GNU/Clang compilers support a memory checking
infrastructure with low performance impact (unlike valgrind's memory
check tool) using the -fsanitize=address flag and linking with the
corresponding libraries. compiling LAMMPS with a compatible compiler
and debug info and then run with this thus instrumented LAMMPS binary
might help to identify memory issues and provides some information
about the location.
another thing you can do to monitor memory usage during a run would be
to periodically execute the "info memory" command. it should give you
a measure of how much memory is being managed by the malloc library on
MPI rank 0. this may have some fluctuations and take some time to
plateau but it should not increase much over time once a plateau is
reached.
5. I currently start simulations from a restart file after "evolving" my cell system to a stable state. Unfortunately I can’t seem to split up the subsequent experiment simulations any further, as without first re-equilibrating the system it instantly crashes (normally with a non-numeric pressure or lost bond/atoms error)
that would indicate that something is not restarting correctly. the
most suspicious piece would be the custom code that is not part of
LAMMPS.
6. I've attached an example python input script and output log file for a (in my case very short) simulation
sadly this is missing the post run information that would be helpful
to identify where in the LAMMPS code time is spend and what degree of
load-imbalance, if any, you are confronted with.
the srun errors at the end are also a bit worrisome. they point toward
some issue or incompatibility with the batch system.
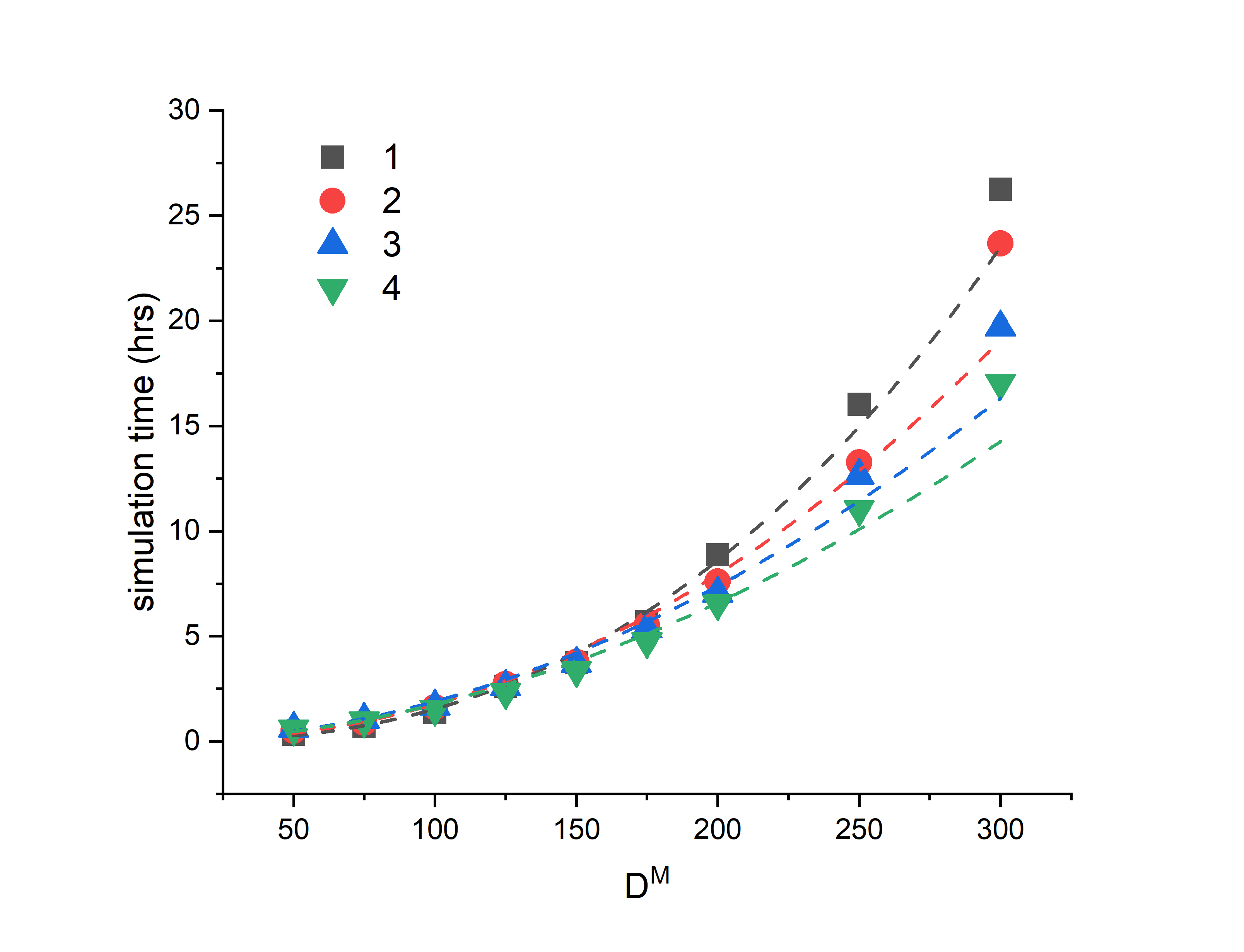
7. The system scales well by increasing the number of CPU's available on a single node, but very poorly when increasing the number of nodes. I've attached a graph of my speed-up testing. The graph shows the total wall-time against cell size (think of D^M as just a total number of particles in the system) for 1-4 nodes of 28 cores. As you can see, at the smaller cell sizes there's next to no speedup (actually slows down in some cases) and only manages to increase marginally at the larger sizes.
scaling performance is mostly independent from whether you have a
single or multiple nodes unless the interconnect between the nodes is
not of a low-latency and high bandwidth kind (like ethernet) or the
MPI library has not been properly compiled/installed/configured to
utilize that hardware.
8/9. It’s a (roughly) homogenous system which I use " fix balance all balance 5000 1.0 shift xyz 20 1.0 weight group 2 water 0.5 bilayer 6.0" To realign processors accordingly. I believe the issue with scaling comes from the use of a custom pair-potential file I use. Testing on boxes of pure lennard-jones water (i.e. without the need for this potential) the system seems to scale much better.
well, that is something that we cannot help you with. for that you
have to debug the custom code or contact its author and ask for
assistance.
axel.