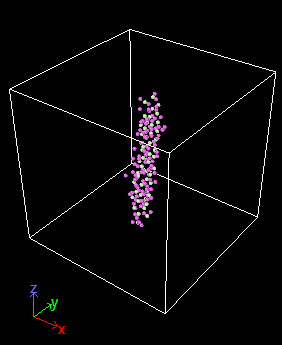
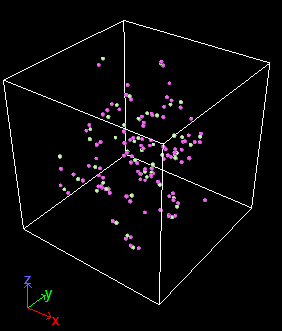
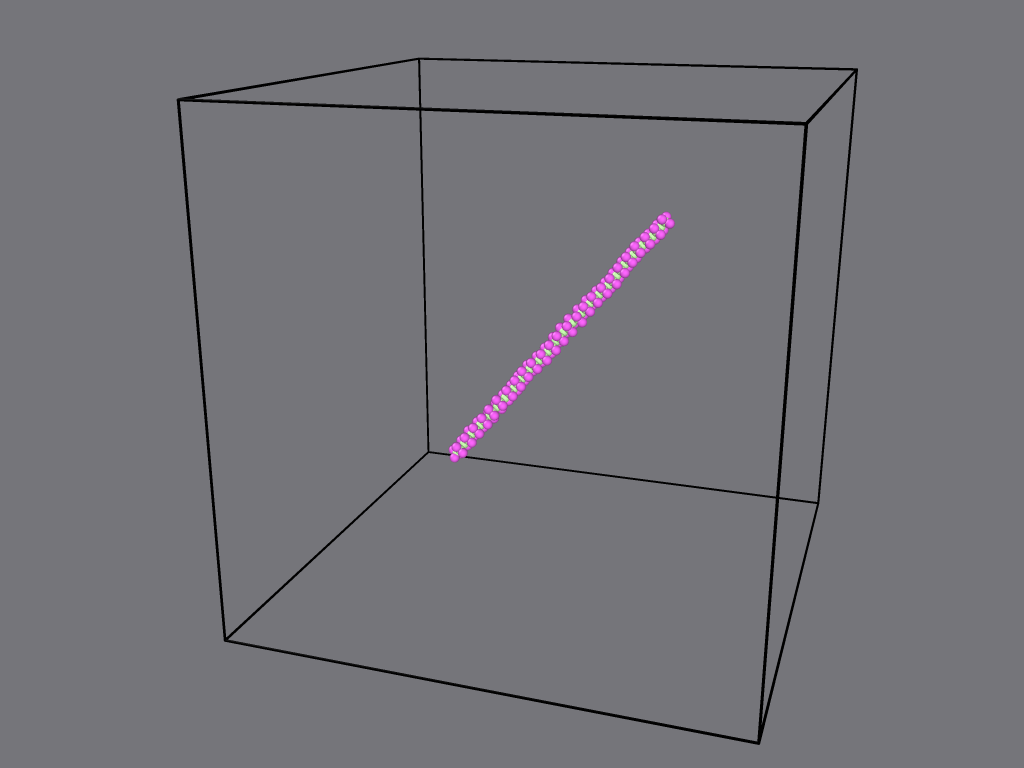
Hello.
I am trying to run a short simulation of polyethylene in a NVT ensemble.
Problem is that PE molecule keeps exploding right after when the simulation starts.
I used a moltemplate example file to model PE. (COMPASS force field)
Code shown below is the LAMMPS code for the simulation.
I would like to know what is the cause of explosion and hopefully, solution too.
Thank you.
units real
dimension 3
boundary p p p
atom_style full
bond_style class2
pair_style lj/class2/coul/long 10.0
angle_style class2
dihedral_style class2
improper_style class2
region box block -50 50 -50 50 -50 50
create_box 45 box bond/types 54 angle/types 113 dihedral/types 382 improper/types 40
include system.in.settings
include PE_mass.data
molecule PEmol PE.mol
create_atoms 0 single 0 0 0 mol PEmol 454756
include system.in.charges
pair_modify mix sixthpower tail yes
special_bonds lj/coul 0.0 0.0 1.0 dihedral yes
kspace_style pppm 0.0001
group all type > 0
min_style cg
minimize 1.0e-4 1.0e-6 1000 10000
timestep 0.2
velocity all create 300.0 12345
fix 1 all nvt temp 300.0 300.0 100.0
thermo 100
thermo_style custom step temp etotal press
dump 1 all atom 500 dump.PE.lammpstrj
run 10000
I don’t see anything in your LAMMPS input that would justify such an explosion. I can only assume that the issue must be originating from the parameter files, but that not something that one can easily review and comment.
What is your LAMMPS version? It must be an old one, since your input does not run with the current version of LAMMPS stopping with an error instead:
ERROR: Molecule auto special bond generation overflow
For more information see https://docs.lammps.org/err0023 (src/molecule.cpp:1557)
This provides the crucial hint at what is missing: your create_box commands has all the “types” settings but no “extra” settings. However those are required or else you get memory corruption or segmentation fault crashes. Once those settings were added, the simulation behaves as should be expected.
Thank you so much! I am using the 64bit 2Apr2025 version. As you said, I added extra settings to solve the error. At the beginning, I created several testing files and during that procedure, I must’ve been uploaded some other faulty code which it wasn’t for my “exploded” molecule.
One last question, is there a method to define the number of “extra/special/per/atom” ? I couldn’t find it by myself so I just put the random number, but I’d like to know how to define it.
It has to be at least the maximum number of special neighbors.
The simplest way to deal with this is to completely avoid them. You write out a data file with your complete initial setup, best with also using the pair ij option so that all pair coefficients are explicitly written out.
Then you can skip loading charges and all coeff commands and thus have a much simpler and faster start using read_data on that data file. This will then automatically determine the required values for all the per/atom storage and apply it. You can use that as a hint for what is needed.
If you read carefully in the documentation how LAMMPS stores bonds, angles, dihedrals, and impropers (depending on the newton bond on/off setting) and special neighbors, you can sort this out yourself. For the most part, you just need to hit the ballpark without being under. Using somewhat larger values only wastes a little bit of RAM.