There was also a bug in molecule.cpp (see attached). Likewise has been updated in the SVN repo.
Paul
molecule.cpp (42.2 KB)
There was also a bug in molecule.cpp (see attached). Likewise has been updated in the SVN repo.
Paul
molecule.cpp (42.2 KB)
Hi Paul,
thanks for the updated routines. The simulation is running now on one core but I cannot say anything about the results yet. When starting the job on more than one core, the simulation crashes immediately after invoking the fix gcmc with the following error-message:
“ERROR on proc 1: Bond atoms 7708 7710 missing on proc 1 at step 1001 (…/neigh_bond.cpp:65)”
Best wishes
Jens
Hi Jens. Your input works for me on 1 or 4 cores, with similar answers between the two. I think that there was a one line change (line 940) in fix_gcmc that made it into the SVN repo yesterday, but not into the version I e-mailed out. Could you try again with the attached and let me know?
Thanks,
Paul
fix_gcmc.cpp (36.5 KB)
molecule.cpp (42.2 KB)
Hi Paul,
the job now also runs for me with different numbers of cores and produces similar numbers of water molecules in the box.
thanks a lot for your help!
Jens
Hi Paul and LAMMPS-Users,
after updating the src-files, the fix gcmc produces molecule numbers that match very well with the gas theory, when inserting only water into a box (independent from the used number of procs). The results are also in excellent agreement with the results from the fix gcmc-molecule written by Sabine Leroch some time ago (see h- and cpp-file attached). However when I apply the two different routines to the insertion of water into a box containing a tio2-nanoparticle, they give very different molecule numbers in the converged state (see attached input-files and diagrams). I talked to Sabine, and she does not know, where this divergence comes from. Her routine is not applying molecule rotations, but if I set the maximum rotation to 0° in the official fix gcmc (maxangle 0) the results are the same as befor. From physical intuition and comparison with experimental results on the water adsorption on tio2, I would prefer the results from Sabines routine, but this is a little bit arbitrary. I will start comparing the two routines, but as I am not very experienced in programming with cpp, I kindly ask, if anyone has a suggestion, which could be the reason for the deviation between the two codes.
Thanks a lot in advance!
Jens
fix_gcmc_molecule.cpp (38.1 KB)
fix_gcmc_molecule.h (2.94 KB)
in.fix_gcmc_particle_-10.9389 (3.06 KB)
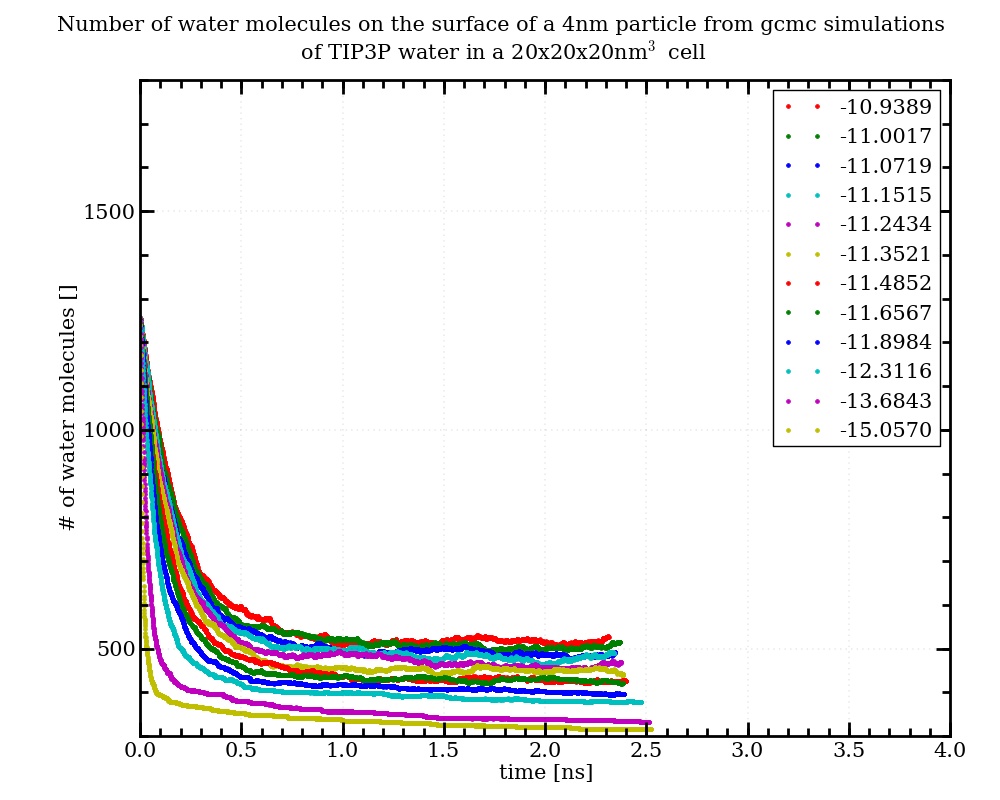
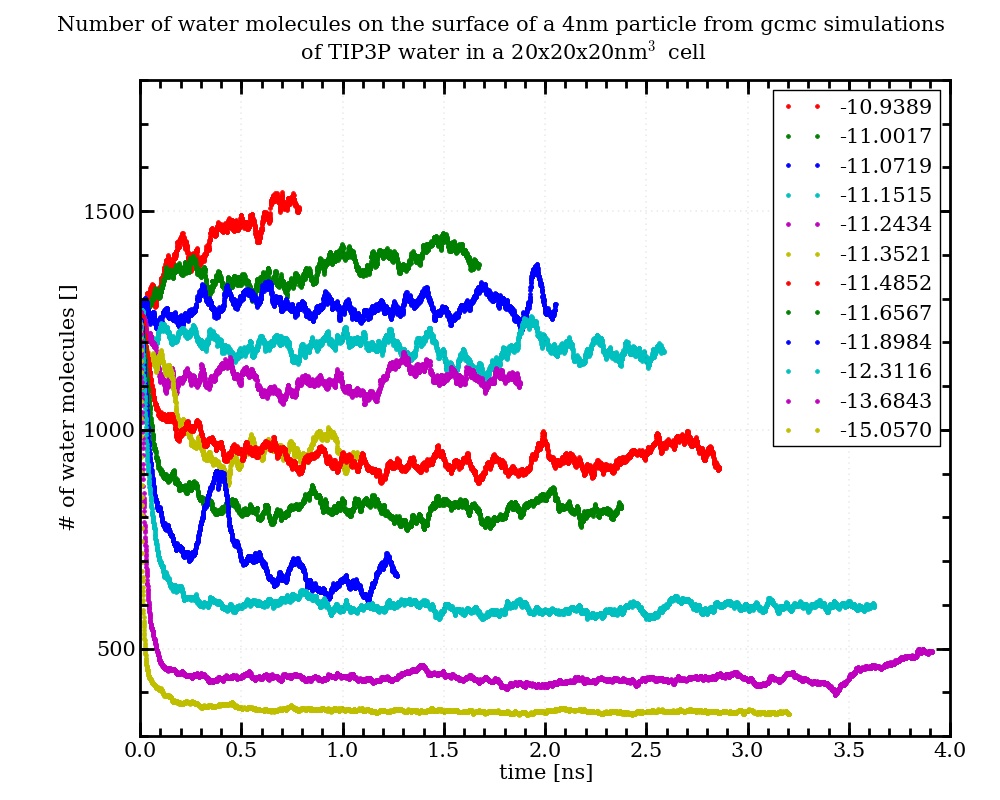
Hi Jens. I haven’t looked into this in detail, but hope to do so soon. Sounds like there is an error in one or both of the versions of fix gcmc, and the error is being demonstrated with this problem. In the meanwhile, please let me know if you have further insight into this.
Thanks,
Paul
Hi Paul,
unfortunately I haven’t found anything significant yet though I am not experienced in debugging. The fix_gcmc_molecule from Sabine is specifically designed to insert water and is based on the official fix_gcmc in the LAMMPS distribution from 13 Dec 2011.
The differences I found so far are the following:
I hope, this helps you at least a little bit.
Best whishes
Jens
Hi Jens. I was able to run the example problem you sent with fix gcmc and I saw the number of waters drop to a low level similar to what you saw. I also tried the same thing with the alternate definition of “zz” that doesn’t include the number of atoms factor, and I still saw similar behavior as you did.
Then I tried running the fix_gcmc_molecule code from Sabine, but ran into hangs and memory issues (probably due to code bugs) with output like this:
Step C_1 TotEng E_pair numberH2 3[1] 3[2] 3[3] 3[4] 3[5] 3[6]
1000 139.76335 -799386.41 -803695.02 1258 0 0 0 0 0 0
ERROR on proc 0: Failed to reallocate 201719808 bytes for array atom:f (…/memory.cpp:66)
application called MPI_Abort(MPI_COMM_WORLD, 1) - process 0
So I’m not able to do direct comparison between the two different versions, unfortunately.
In looking at the source code, I did notice that the details of how the molecule energies are computed are considerably different, so the difference probably lies there. It is possible that there’s a problem with the input you sent, but there’s likely at least one error in one or both of these versions of the fix gcmc code when running this particular problem.
I’d recommend outputting the computed insertion and deletion energy distributions and Boltzmann factors for both versions and comparing them. That might shed some light on what is going on here.
Best wishes,
Paul