Feature request: Perhaps at the start of a run, LAMMPS could scan through the list of bonded pairs of atoms and print out an error message if any pair of atoms exceeds the comm distance threshold? (And indicate what the distance was between the offending pair of atoms.)
Although this is mentioned in the documentation, and has come up in past discussions on the mailing list. It’s happened to me 3 times over the years, just infrequently enough that I forget what the problem is.
Meanwhile, I’m posting a short rant here to increase the odds a google search will take people quickly to the solution:
If you are running a simulation and pairs of atoms straddling the periodic boundaries behave as if their bonds are stretched across the simulation box (and are attached to the wrong periodic image), this means you need to use the “comm_modify cutoff” (or “comm_modify multi/cutoff”) command to increase the communication distance. It must exceed the maximum length of the bonds in your system. In the image below, one of the pairs of bonded atoms was initially slightly longer than the other, exceeding the default comm distance, and the result was:
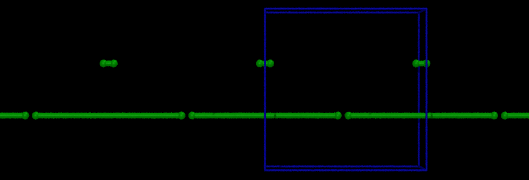
(two identical bonds shown, including their periodic images, after equilibration)
This is only one of the kinds of mayhem that can result from failing to increase the comm cutoff. It’s not always so obvious visually. (In some cases, I have to step through the code with a debugger to figure out what went wrong.)
If LAMMPS can halt and print out a message indicating how large to set the comm distance cutoff, it would be extremely helpful.
Thanks for your time, and please forgive the rant.
Andrew
P.S. I admit this tends to happen more often to people running coarse grained simulations. All-atom simulations might not run into this problem since the bond lengths are less than the largest heavy atom diameters which determine the default comm distance.