Hi everybody,
The force field parameters in ffield.reax.lg seems wrong.
Does anybody have the correct file of that?
I mean the force field used in the following paper “# ReaxFF-lg: Correction of the ReaxFF Reactive Force Field for London Dispersion, with Applications to the Equations of State for Energetic Materials”
I really appreciate your help.
Regards,
Sara
Please explain why you think the provided reax.lg file
is wrong?
Steve
Dear Steve,
I was not able to run with this force field but I run the sucrose crystal with other force field provided in the potential force field.
I get the below error

I attached my input file, the data file and the force field. My lammps version is 14 Feb 2018.
I appreciate your reply.
Kind regards,
Sara
data.sucrose333c (118 KB)
ffield.reax.lg (15.1 KB)
in.sucrose333 (2.11 KB)
Dear Steve,
I was not able to run with this force field but I run the sucrose crystal with other force field provided in the potential force field.
I get the below error

I attached my input file, the data file and the force field. My lammps version is 14 Feb 2018.
this is NOT an indication that the force field file is incorrect, but a sign of a problem of your input.
the .lg reaxff force field file has a different format than the regular reaxff force field files and thus you need to set a required keyword on the pair_coeff line. please see the manual for details.
i would also recommend upgrading to a newer version of LAMMPS, which has some relevant bugs fixed and a better detection for user input errors like this.
axel.
Hi,
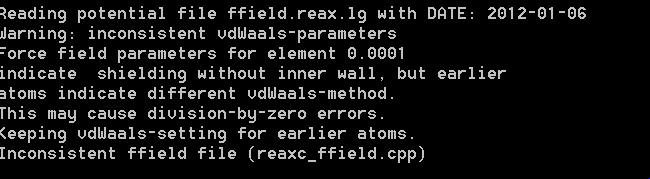
I finally run my crystal with ffield.reax.lg but I have some question about it.
After running, in the error file, there is an error. The file is attached.
I simulated a technical report by Scott Weingarten (attached). I would like to know what is the reason for the difference between my results and his. I read the manual, it says " Using the optional keyword lgvdw with the value yes turns on the low-gradient correction of the ReaxFF/C for long-range London Dispersion, as described in the (Liu) paper. Force field file ffield.reax.lg is designed for this correction, and is trained for several energetic materials (see “Liu”). When using lg-correction, recommended value for parameter thb is 0.01, which can be set in the control file."
I simulate the crystal with lmp_conrtol file (attached) and with default values from reax/c (NULL). I summarised my results in below table.
with keyword NULL the calculation seems better but not exactly the same. Would you please tell me if the amount of thb=0.01 is embedded in the force field? also is there any reason that my results are not exactly the same?
|
Reported in the report
|
pair_style reax/c NULL lgvdw yes
|
pair_style reax/c lmp_control lgvdw yes
|
- | - | - | - |
a
|
11.128
|
10.84
|
8.62
|
b
|
8.771
|
8.59
|
8.91
|
c
|
7.784
|
7.94
|
9.23
|
α
|
89.02
|
91.10
|
87.42
|
β
|
105.83
|
99.10
|
106.11
|
ɣ
|
91.42
|
90.04
|
92.61
|
density
( g/cm3)
|
1.56
|
1.54
|
1.60
|
Regards,
Sara
data.sucrose333c (118 KB)
ffield.reax.lg (15.9 KB)
in.sucrose333 (2.07 KB)
lmp_control (1.06 KB)
sucrose.e9377182 (509 Bytes)
ARL-TN-0666.pdf (864 KB)
The default values for thb and other parameters can be found here: https://lammps.sandia.gov/doc/pair_reaxc.html
Have you considered the standard deviation of your results? Your simulation seems too short to me – it’s only about 5 ps.
Michal
Hi Michael,
The simulation time is around 50 ps. I do know where the default parameters in Lammps document are. The thing that I do not know the value of the thb when I use NULL in my simulation.
Thank you
Sara
Unless I’m missing something, your simulation consists of 47500 steps with a timestep of 0.1 fs, which gives 4750 fs=4.75 ps. If you use NULL, the default values for parameters are used.
Michal
It may not be obvious, but “thb” is “thb_cutoff” – so the default value is 0.001.
Hi Ray,
Would you please confirm that when I use NULL for all the ffield.reax the default parameters which are mentioned in the manual are used or the default parameters for each force field are different???
The other question of mine is that I kept all the default parameters in the manual unchanged and just change the value of “thb_cutoff” to 0.01 as manual said for ffield.reax.lg. But as I showed in the table in my first e-mail the unit cell parameters are again different from what is reported in the paper by Weingarten. I think the calculated parameters are closer to what is reported with NULL. Would you please comment on this.
Kind regards,
Sara