Hello GULP team,
In GULP manual, “If a shell model has been specified for a given atom in the species section, but the shells are omitted from the input for the structure then GULP will
attempt to add them automatically. When this happens, the shell be placed at the
same location as the core by default.” But when I do that, the result shows that it
only takes core coordinates into consideration,which makes my system not charge-neutral.
my version is 6.2.
I want to know the reason.Thanks in advance!
Xiaohan
It would help if you could post your input file please, otherwise it’s hard to guess what’s wrong.
Thanks,
Julian
Sorry,it was my carelessness.These are my input and output files.Thanks in advanced!
PZO_corrected.gin (2.8 KB)
output1.gin (841 Bytes)
Thanks for sharing your input. I’ve investigated and there was a couple of hiccups in the code for your case. If you want to patch your code then here are the fixes for version 6.1 that you are using:
- In setcfg.F90 at line 381: Insert a line below this (i.e. in the loop) which is:
ncf = i - In addshell.F90 at line 101: Change this line to:
lshellneeded = ((natspec(nshellspec)-ni).eq.maxele.and.(ntypi.eq.ntypspec(nshellspec).or.ntypspec(nshellspec).eq.0))
The shells will then be added. However, note that something is wrong with your 2nd structure in that the charges don’t quite add up correctly & so you’ll need to sort this out before you can run your input.
Regards,
Julian
Sorry to bother you again,there are still some issues.
addshell.F90 at line 101:do while (nshellspec.lt.nspec.and…not.lshellneeded).chage this line?
At line 103:lshellneeded=((natspec(nshellspec)-ni).eq.maxele.and.ntypi.eq.ntypspec(nshellspec)).I think I maybe should modify the line 103.But the result shows that the system still hasn’t automatically added the shell.
This is what the code in that region should look like now:
lshellneeded = .false.
nshellspec = 0
do while (nshellspec.lt.nspec.and..not.lshellneeded)
nshellspec = nshellspec + 1
lshellneeded = ((natspec(nshellspec)-ni).eq.maxele.and.(ntypi.eq.ntypspec(nshellspec).or.ntypspec(nshellspec).eq.0))
enddo
Hope that clarifies things.
Now the system can automatically add shells.Thank you very much!
But it can’t recognize the atoms which have different names from the species definition.
Sorry to bother you again!
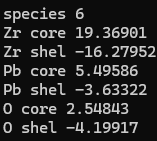
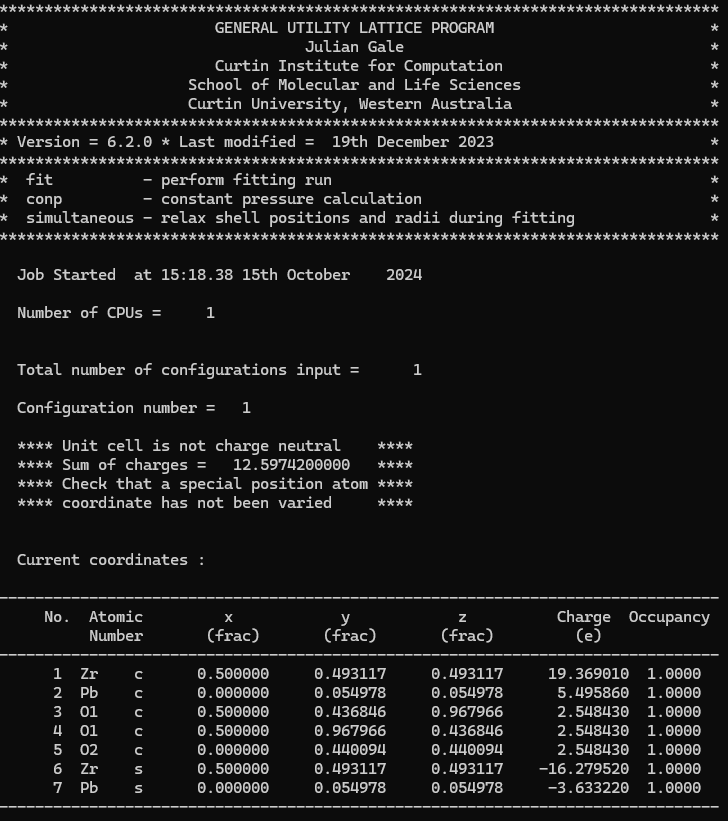
I’m not sure what’s happening in your input/version. I’ll attach an example of input and output that shows that shells are added (based on one of your input structures). This is for 6.2.3 (which is available for download, so there is no need to modify the code for the fix). The run stops with an error, but this is just a rounding error in the charges leading to a small deviation from neutrality.
addshells.got (8.0 KB)
addshells.gin (799 Bytes)
I have run the input file you provided,but the result is different from yours.I feel
that the system doesn’t think O1 belongs to the O.
Hope you can tell me the solution.I’m really sorry to disturb you so many times.
addshells.got (5.5 KB)
The solution is actually in my previous post - if you install 6.2.3 (my version of the patched code) then you’ll see exactly the same results as me.
Dear Julian,
I downloaded the latest version and the issue has been resolved.Thank you for your patient guidance!
Xiaohan