I am simulating a FeCr system with a single dumbbell interstitial defect at 700K. For tracking the Fe/Cr atoms I am using ‘compute displace/atom’ and printing it to a file using ‘fix ave/time’. I am comparing the printed displacement values with the dump file in Ovito. The values match up perfectly fine.
However, if in the same sim I keep the Cr at 0 (pure Fe system), the values do not match up, as if the atoms are rigid.
Attached is a simplified version (with dumb_int6.lammps (3.3 KB) FeCr_d.eam.alloy (692.5 KB)
no defect) of my script along with the potential file. It should work with LAMMPS compiled with the ‘basic’ preset. To reproduce the results you just need to change line 59 where I specify the number of Cr atoms. First make it 1 and then make it 0 and compare at the Fe_run1.dat file in both the cases.
And if you just want to have a quick glance at the script, following:
Note: Notice, at 0% Cr, the magnitude of displacement is correct but not the individual dx, dy and dz.
clear
variable run index 1
variable rand index 579482
variable all_dn string 'all_run1.dump'
variable dn string 'run1.dump'
variable dnFe string 'Fe_run1.dat'
variable dnCr string 'Cr_run1.dat'
##################################################
# Setting up simulation conditions
##################################################
label reload
units metal
dimension 3
boundary p p p
atom_style atomic
atom_modify map array
###################################################
# Creating a BCC lattice
###################################################
variable lat_param equal 2.85532 #initial lattice param
variable cell_len equal 10 #number of lattices
lattice bcc ${lat_param} orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
region box block 0 ${cell_len} 0 ${cell_len} 0 ${cell_len} units lattice
create_box 2 box
create_atoms 1 box
#masses
mass 1 55.845 #Fe
mass 2 51.9961 #Cr
variable tstep equal 100
thermo ${tstep}
thermo_style custom step time temp pxx pyy pzz lx ly lz
###################################################
# Pair Potential, Calculations and I Minimization
###################################################
pair_style eam/alloy
pair_coeff * * FeCr_d.eam.alloy Fe Fe
neighbor 2.0 bin
neigh_modify delay 10 check yes
fix 1 all box/relax aniso 0.0 vmax 0.001 ##experiment
min_style cg
minimize 1e-16 1e-16 20000 30000
unfix 1
######################################################
# Adding Cr to the Fe system and II Minimization
######################################################
variable numCr equal round(1) #no. of Cr atoms
set type 1 type/subset 2 ${numCr} ${rand}
fix 1 all box/relax aniso 0.0 vmax 0.001 ##experiment
# II min. : after SOLUTES HAVE ARRIVED (cue entrance music)
min_style cg
minimize 1e-16 1e-16 20000 30000
unfix 1
###################################################
# ADDING HEAT
###################################################
reset_timestep 0
variable simT equal 700
velocity all create ${simT} ${rand} mom yes rot yes
fix RC all recenter INIT INIT INIT
fix NPT all npt temp ${simT} ${simT} $(10.0*dt) aniso 0 0 $(1000.0*dt)
fix NVE all nve
fix MOM all momentum 1000 linear 1 1 1
run 1000
unfix RC
unfix NPT
unfix NVE
unfix MOM
###################################################
# PRODUCTION
###################################################
reset_timestep 0
group grp_Fe type 1
group grp_Cr type 2
#atom count
variable Fe_atoms equal count(grp_Fe)
variable Cr_atoms equal count(grp_Cr)
#displacement computes
compute disp_Fe grp_Fe displace/atom
compute disp_Cr grp_Cr displace/atom
compute disp_Fe_x grp_Fe reduce sum c_disp_Fe[1]
compute disp_Fe_y grp_Fe reduce sum c_disp_Fe[2]
compute disp_Fe_z grp_Fe reduce sum c_disp_Fe[3]
compute disp_Fe_mag grp_Fe reduce sum c_disp_Fe[4]
compute disp_Cr_x grp_Cr reduce sum c_disp_Cr[1]
compute disp_Cr_y grp_Cr reduce sum c_disp_Cr[2]
compute disp_Cr_z grp_Cr reduce sum c_disp_Cr[3]
compute disp_Cr_mag grp_Cr reduce sum c_disp_Cr[4]
dump sys all custom 10000 ${all_dn} id type x y z
fix RC all recenter INIT INIT INIT
fix NVE all nve
fix MOM all momentum 1000 linear 1 1 1
fix Fe_track all ave/time 1 1 ${tstep} c_disp_Fe_x c_disp_Fe_y c_disp_Fe_z c_disp_Fe_mag v_Fe_atoms file ${dnFe}
fix Cr_track all ave/time 1 1 ${tstep} c_disp_Cr_x c_disp_Cr_y c_disp_Cr_z c_disp_Cr_mag v_Cr_atoms file ${dnCr}
run 10000
undump sys
print "We won soldier!"
First off, if you quote your input file, you should quote it correctly by using triple backquotes (```). Have a look yourself and see that parts of it are rendered in an unintelligible fashion.
There are two major problems with your input and how you compare results:
Compute displace/atom considers atoms passing through periodic boundaries, but your dump file only storing the positions for the periodic copy in the simulation box (x, y, z) instead of unwrapped coordinates (xu, yu, zu) or regular coordinates and image flags (x, y, z, ix, iy, iz). That will lead to discrepancies if you have significant reconstruction and thus atoms moving through periodic boundaries.
Your use of fix recenter invalidates the computed displacement values since it will result in removing any center of mass motion from the displacement vectors.
I fixed the triple backquotes, thank you for that Axel.
but your dump file only storing the positions for the periodic copy in the simulation box (x, y, z) instead of unwrapped coordinates (xu, yu, zu)
In the post-processing, before comparing the displacements (with LAMMPS output) I do unwrap the boundaries in Ovito. The difference in output is significant when Cr is 0, the LAMMPS reports displacement as if the atoms are rigid.
Your use of fix recenter invalidates the computed displacement
I am using this to get rid of any drift in the system, this does not affect the calculations/dynamics.
To sum up, when Cr is 0% in the system, the displacements reported via LAMMPS are of rigid atoms. However, if there is even a single atom of Cr, the reported displacements look reasonable and also match with post-processed dump files.
You’re not initiating the system with any momentum velocity and the integrators you use do not add any drift. Your use of fix recenter and momentum are not needed and disturb the dynamics.
Speaking of which, in your MWE file I can read the following:
fix RC all recenter INIT INIT INIT
fix NPT all npt temp ${simT} ${simT} $(10.0*dt) aniso 0 0 $(1000.0*dt)
fix NVE all nve
fix MOM all momentum 1000 linear 1 1 1
This is bogus. One integrator should be enough. Please read the fix nvt or fix nve dedicated sections of the manual. There are also warning messages in the log file.
Your example file sets 1 atom to type 2 (Cr). This does not correspond to the situation you are describing. If remove line 61 which sets some atoms to type 2, I get a displacement of 0 from type 2 atoms.
This sentence makes no sense. The displacement value of an empty group will be 0 as the value is set to 0 for atoms not in the group. This does not means that the atoms are rigid. Please read the documentation of the compute displace/atoms command.
If you start from an ideal crystal geometry and are below the melting point, why should there be a significant motion, particularly when you suppress even fluctuations around the center of mass with fix recenter?
Mind you, I also just noticed that your equilbration is bogus because you are using two time integrating fixes at the same time. Didn’t LAMMPS print a warning about that?
If there would be a drift in your system even with using fix momentum, then there is something seriously wrong with your simulation.
While fix recenter does indeed not change the dynamics, it does change the frame of reference. Yet compute displace/atom requires that all displacements are viewed from the frame of reference of the first step where the compute is active. So the computed result must be bogus or fix recenter would not have a significant impact and thus would be useless.
That said, if you believe that the output of fix ave/time is incorrect, you need to provide proper proof, e.g. by using a different method of output, e.g. via thermo_style custom or fix print. That you don’t get the expected result is no proof for a failure of fix ave/time, but could just as well be cause by a simulation setup that is not what you assume it is.
I got two ‘bogus’ in a row, I ought to be attentive from here on.
My actual simulation has defects, and runs for 250 million timesteps. Due to numerical error accumulating I start to see some drift after ~125 million timesteps. In fact, I did ask this question earlier in a different post. When I posted the simplified script for the forum with only a few thousand timesteps, I should have removed the ‘fix recenter’. I have removed it now (attached script).
Completely agree, what was I thinking? I apologize, I should have been more careful.
I am attaching two log files log_Cr0atoms.lammps and log_Cr1atoms.lammps which are the log files where I am printing displacement of Fe atoms using ‘thermo_style custom’ when number of Cr atoms is 0 and number of Cr atoms is 1, respectively. Notice, for the same simulation, when number of Cr atoms is 0, the total displacement of Fe atoms (x, y, z) is like rigid. However, if I add 1 Cr atom to the system the total displacement of Fe atoms (x, y, z) is some reasonable value (compared to rigid values).
I have responded to your questions (and comments) in the reply to Axel, and I agree my equilibration was bogus. I should be careful. About the displacement, I am referring to the displacement of Fe atoms and not Cr. Of course, displacement will be zero with 0 Cr atoms in the system.
I am attaching two log files log_Cr0atoms.lammps and log_Cr1atoms.lammps which are the log files where I am printing displacement of Fe atoms using ‘thermo_style custom’ when number of Cr atoms is 0 and number of Cr atoms is 1, respectively. Notice, for the same simulation, when number of Cr atoms is 0, the total displacement of Fe atoms (x, y, z) is like rigid. However, if I add 1 Cr atom to the system the total displacement of Fe atoms (x, y, z) is some reasonable value (compared to rigid values).
Ok so I misunderstood your issue. Let me answer this with a simple question: What do you think the average displacement of a box with zero total momentum will be, if you compute it over all the atoms in the simulation box?
I completely understand your point, but since there is a non-zero temperature, the atoms will still jiggle. This means the net displacement will be close to zero and not exactly zero. For instance, compare the log files I shared while replying to Axel. The displacement reported for log_Cr1atoms.lammps is close to zero, but the displacements in log_Cr0atoms.lammps are exactly(ish) zero.
You would also notice, the magnitude of displacement in the log_Cr0atoms.lammps does not match up with the sum of dx or dy or dz.
Now, if you do a quick post-processing of the dump files in both cases, the Cr1 cases match up just fine, but Cr0 cases does not.
This is exactly what my remark is concerned with. Please reconsider it. What value do you get when computing the difference between your 1 Cr atom displacement and all you Fe atoms displacement? Do it in LAMMPS, draw some conclusion.
There is not cause-consequence relation between these two assumptions. This depends on the equations of motion you integrate and initial conditions of your system. Also the finite value of the center-of-mass displacement can appear from rounding errors and oscillate around a negligible drift value.
Yes. This is because vector magnitude are not signed. Displacements are. This is basic algebra as the magnitude is computed with |V|=sqrt(V_{1}^{2}+V_{2}^{2}+V_{3}^2). If you sum the displacement magnitude on a lot of atoms, you will get a big number even if the displacement of each one is small.
Do not assume people will do the same “quick post-processing” as you do. If you want people to comment on your results, tell them which tools you use, what you compute and how. As for now I see no issue with the displace/atom command outputs so I still do not understand what your issue is and where you’re going to with your remarks.
I might be misphrasing my problem constantly. Alright, let me give this another shot (I got this!).
In my actual simulation, there are defects within the system that ultimately lead to the diffusion of atoms. However, for easy reproduction of the results, I removed that defect and shared an elementary LAMMPS script with just heating
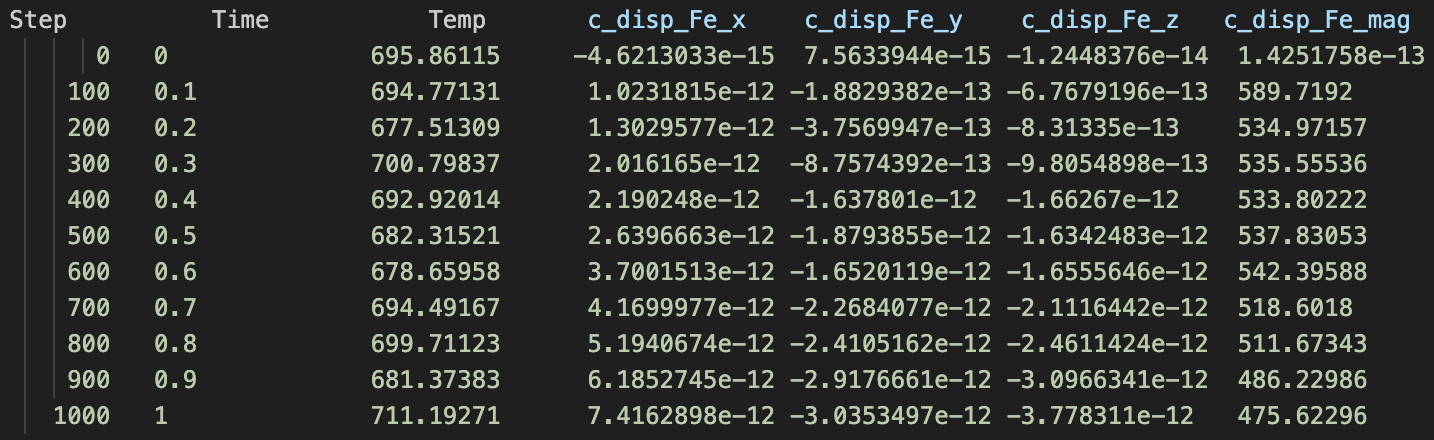
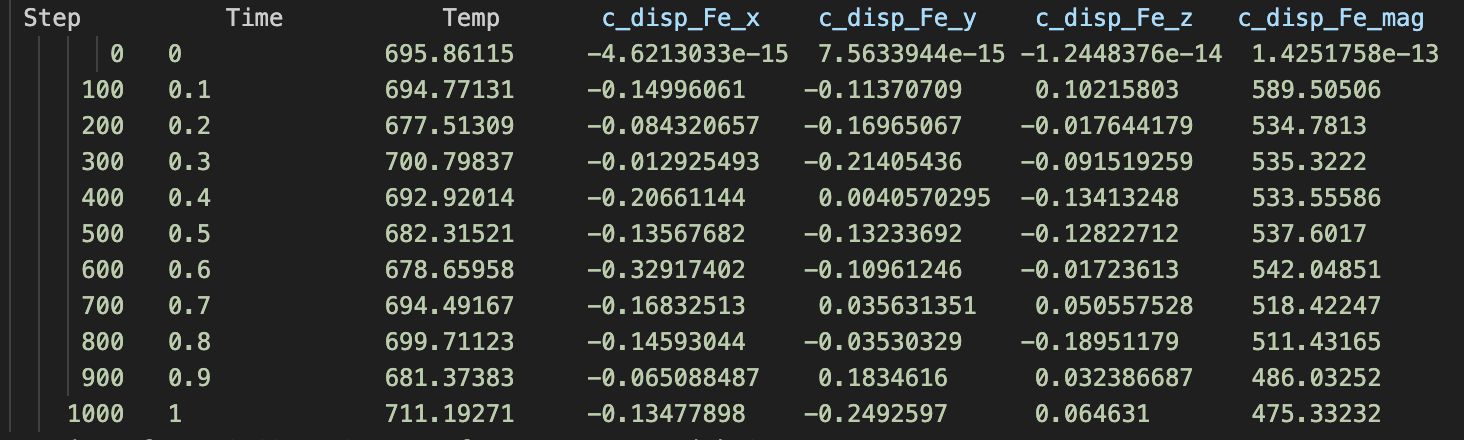
A system with 2000 Fe atoms (pure Fe) [let’s call this sim1]
A system with 1999 Fe atoms and 1 Cr atom [let’s call this sim2]
During each of the simulations, I am printing sum of displacements in the x, y and, z directions for Fe atoms only, and let’s call this disp_Fe_x,y,z. The problem is, that the values are different. How different you ask? In sim1 the disp_Fe_x,y,z values are of the order of magnitude 1e-13, while in sim2, the same values are ~0.1. I have attached the screenshot of the log files. So my question is, why this mismatch? To reiterate, the only difference between sim1 and sim2 is 1 Cr atom in the system.
Now about the postprocessing, the script I shared also prints a dump file. If you open it in Ovito (or any of your visualization software) and and see the displacement between the first frame and the last, you would notice that disp_Fe_x or disp_Fe_y or disp_Fe_z is not of the order 1e-13 in the case of sim1.
When a system of identical-mass particles follows equations of motion that conserve momentum, its total displacement should be zero for all time in any frame where its initial total velocity was zero.
That makes sense to me, and I think I can connect that with Germain’s response, ‘cause-consequence relation’.
In my actual problem, I am trying to simulate a single dumbbell interstitial defect in a crystal lattice (pure Fe) at 700K. The simulation runs for 100 mil. timesteps (with fix NVE) leading to the diffusion of Fe atoms. As the simulation runs, I want to track the sum of displacement of Fe atoms in x, y and z directions separately. Currently, the sim is printing 0 displacements even after 100 mils. timesteps. Is there a way around this?
When a system of identical-mass particles follows equations of motion that conserve three-dimensional momentum, its total displacement in any direction should be zero for all time in any frame where its initial total velocity was zero.