Hello LAMMPS community,
I am running LAMMPS 22 Jul 2025.
I am trying to simulate a coarse-grained model of crystal formation using fix bond/react (Tagging @jrgissing for your expertise). In this model, each molecule is a trimer with 3 binding sites. I am implementing both “forward” (association of a trimer) and “backward” (dissociation) reactions. For now my molecules are rigid bodies (fix rigid).
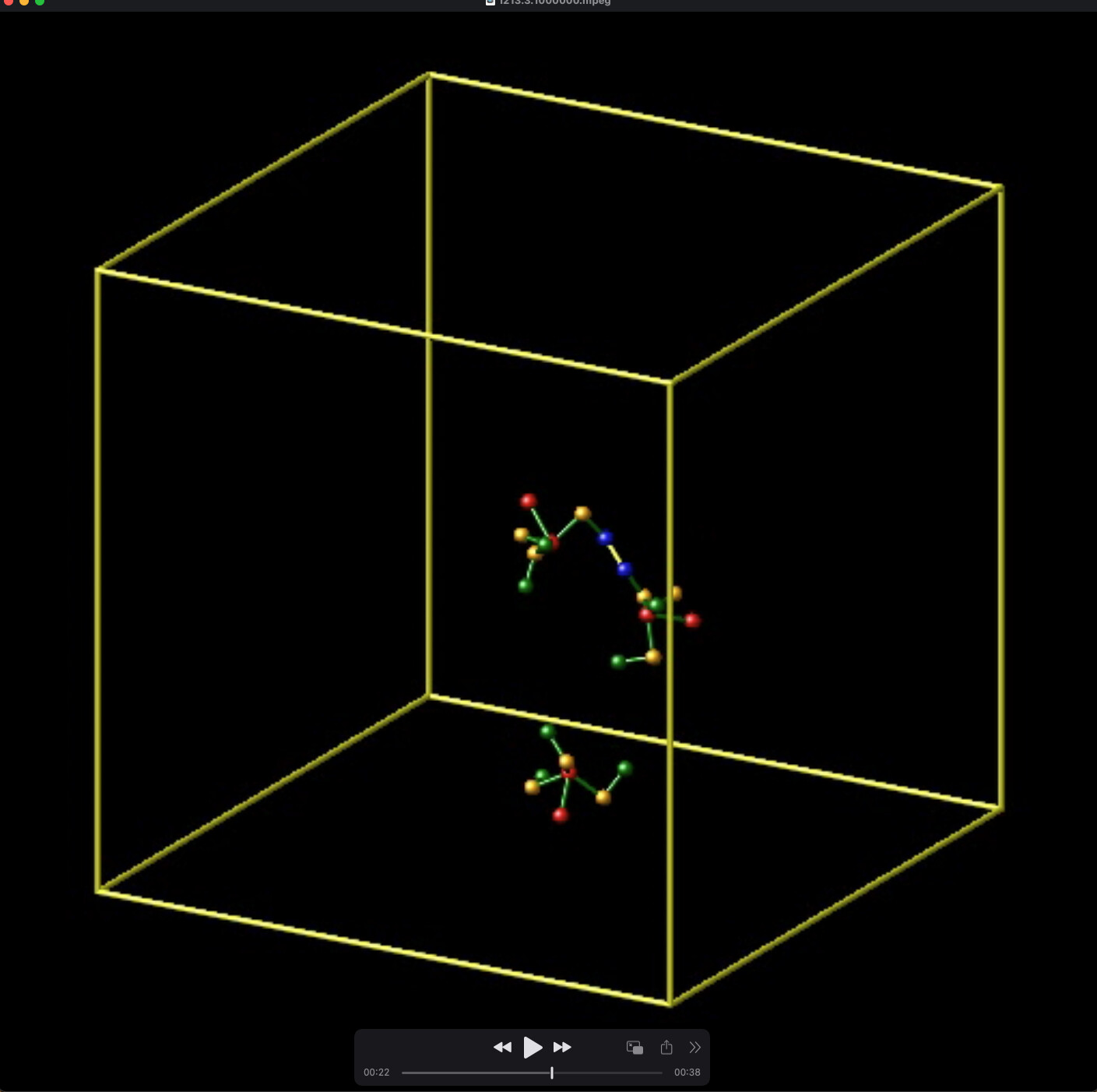
I initialized the simulation with 3 trimers that are bonded linearly, say, mol 2-1-3 (Screenshot 1). When one trimer dissociates, say mol 2, a dihedral property between mol 1-3 is removed erroneously. In my design, each bond has 3 dihedrals. However, the data file shows that after dissociation of one trimer, there are only 2 dihedrals – the one missing is between atoms 2,4,7,22. This results in the remaining bonded molecule pairs “flopping” around (Screenshot 2).
My suspicion is that this deletion is due to edge atoms being too close to each others and the three reaction sites being partially overlapping.
My questions are:
- Why does this happen? I thought that with dihedrals, as long as I include atoms within 3 bonds of the edge atoms, the simulation should be fine (no errors nor warnings were generated indeed).
- I do not want to define reaction templates and maps for different occupancy status of the trimers. Is it enough to increase the number of atoms in the “arms” (see annotation in molecule templates) from 2 to 3?
Thank you so much in advance for your time and help!
Best,
Richard
Screenshot 1: 3 trimers bonded in intended configurations
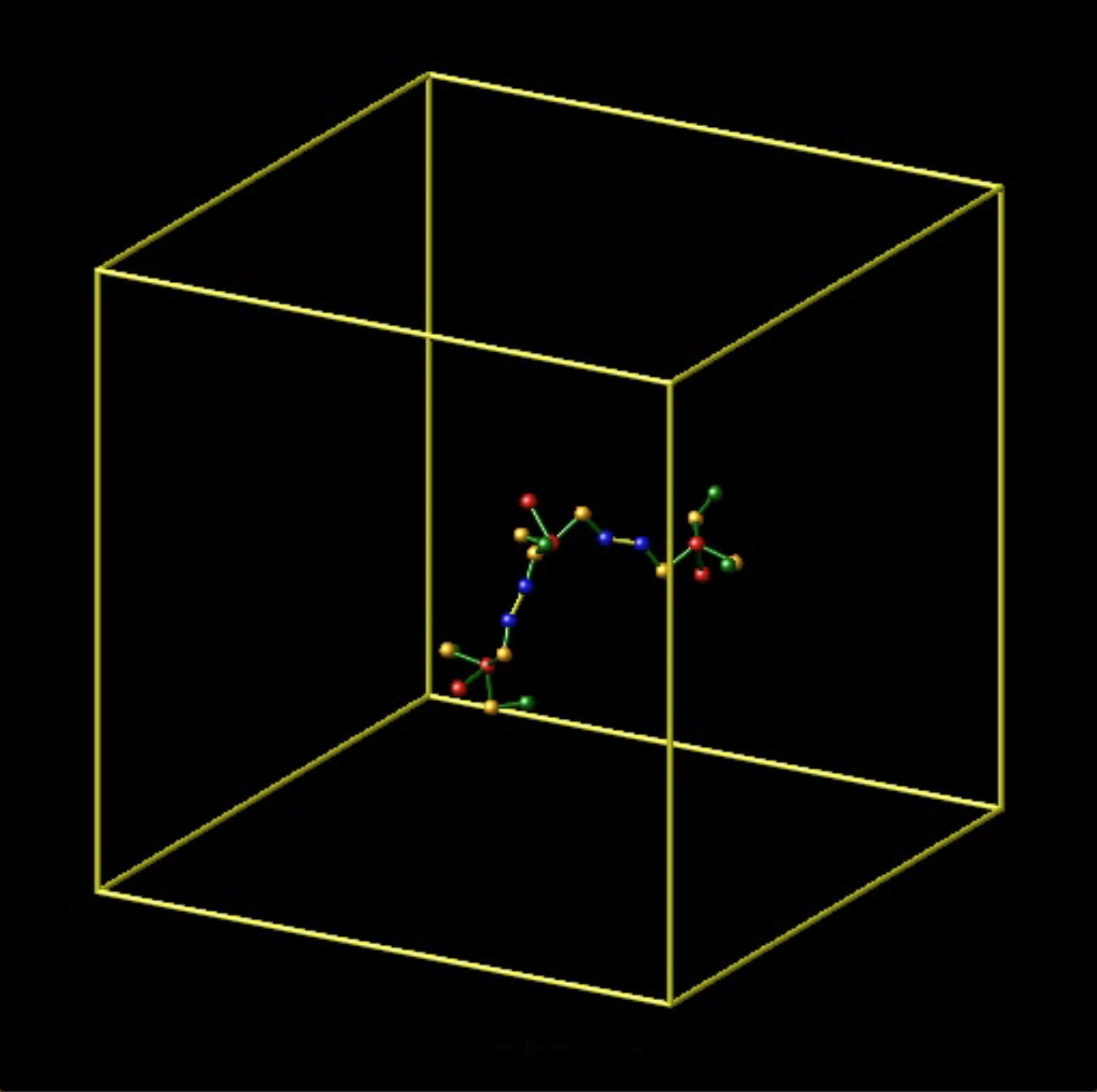
Screenshot 2: After a trimer dissociates, a dihedral angle of the remaining bond is erroneously removed.
Below are the files relevant to the in file, initial configuration file, output data file
in.I213.3-3.1000000.txt
units nano
boundary p p p
atom_style molecular
pair_style hybrid lj/cut 6.6 zero 6.6
bond_style harmonic
angle_style cosine/shift/exp
dihedral_style cosine/shift/exp
read_data Out/I213.3.dat &
extra/special/per/atom 10 extra/bond/per/atom 3 &
extra/angle/per/atom 1 extra/dihedral/per/atom 1
pair_coeff 1 1 lj/cut 4.14 3.0 3.3674 # 1.5
pair_coeff 1 2 lj/cut 4.14 2.6 2.9184 # 1.5 + 1.1
pair_coeff 1 3 lj/cut 4.14 2.6 2.9184 # 1.5 + 1.1
pair_coeff 1 4 lj/cut 4.14 2.6 2.9184 # 1.5 + 1.1
pair_coeff 2 2 lj/cut 4.14 2.2 2.4694 # 1.1
pair_coeff 2 3 lj/cut 4.14 2.2 2.4694 # 1.1
pair_coeff 2 4 lj/cut 4.14 2.2 2.4694 # 1.1
# pair_coeff 3 3 lj/cut 4.14 2.2 6.6 # 1.1 # 3s
pair_coeff 3 3 lj/cut 4.14 2.2 2.4694 # 1.1 # 3s
pair_coeff 3 4 lj/cut 4.14 2.2 2.4694 # 1.1
pair_coeff 4 4 lj/cut 4.14 2.2 2.4694 # 1.1
pair_coeff 4 4 zero # 1.1
pair_modify shift yes
special_bonds lj 0.0 1.0 1.0
region bbox block EDGE EDGE EDGE EDGE EDGE EDGE
fix bbox_reflect all wall/region bbox harmonic 41.4 0 3
# molecule I213 I213.mol.txt
# region core block -20 20 -20 20 -20 20
# create_atoms 0 random 5 1000021 core mol I213 1000001
# fix prop all property/atom mol
variable cluster atom ((id-1)/8)+1
set atom * mol v_cluster
group mobile id > 8
fix 1 mobile rigid molecule langevin 300 300 1 3674340
# fix 1 all rigid molecule langevin 300 300 1 3674342
# neigh_modify exclude molecule/intra all
# neighbor 5 bin
# neigh_modify every 1 delay 0
molecule mol1 rxn1_pre.txt
molecule mol2 rxn1_post.txt
fix myrxns all bond/react stabilization no &
react rxn1 all 1 2.3 2.4 mol1 mol2 rxn1_map.txt &
react rxn2 all 1 2.56 3.0 mol2 mol1 rxn1_map.txt
# fix 1 all nve
# fix 2 all langevin 300 300 1 3674339 zero no
timestep 0.01
thermo 100000
thermo_style custom step temp press density f_myrxns[1] f_myrxns[2]
# thermo_style custom step temp press density
dump 1 all movie 1000 Out/I213.3.1000000.mpeg &
type type bond type 0.3 zoom 1.2 view 70 30 box yes 0.01
dump_modify 1 acolor 1 red acolor 2 orange acolor 3 green acolor 4 blue
dump_modify 1 bcolor 1 green bcolor 2 green bcolor 3 green bcolor 4 yellow
dump_modify 1 adiam 1 1.0 adiam 2 1.0 adiam 3 1.0 adiam 4 1.0
# dump_modify 1 adiam 1 3.0 adiam 2 2.2 adiam 3 2.2 adiam 4 2.2
dump 2 all custom 10000 Out/I213.3.1000000.dump id mol type xu yu zu
dump_modify 2 sort id
# restart 1000000 Out/I213_Index_1.restart
run 900000
# unfix bbox_reflect
run 10000
write_data Out/I213.3.1000000.dat nofix
Out/I213.3 dat (initial configuration file read by read_data)
LAMMPS data file via write_data, version 22 Jul 2025, timestep = 10000, units = nano
24 atoms
4 atom types
23 bonds
4 bond types
4 angles
1 angle types
6 dihedrals
2 dihedral types
-20 20 xlo xhi
-20 20 ylo yhi
-20 20 zlo zhi
Masses
1 9.42478
2 9.42478
3 9.42478
4 9.42478
Bond Coeffs # harmonic
1 828 3
2 828 2.6
3 828 2.2
4 828 2.3439
Angle Coeffs # cosine/shift/exp
1 828 138.546 20
Dihedral Coeffs # cosine/shift/exp
1 828 169.821 200
2 828 -176.209 200
Atoms # molecular
12 1 2 1.590802376771 -3.0698719289422507 -10.987164817646569 0 0 0
9 1 1 4.2637315345706215 -3.720737187049676 -8.92427604654249 0 0 0
15 1 3 0.04311206086622721 -1.5101483282358172 -10.877999847313177 0 0 0
16 1 3 0.1100499383024558 -6.756175524851395 -8.69975026083126 0 0 0
13 1 2 1.9496188256827622 -5.987057055548208 -7.769995936263177 0 0 0
10 1 1 1.3057317825933021 -3.5615675592248035 -8.450037268897582 0 0 0
11 1 2 2.3173994833615943 -1.7321985203314667 -6.904080148571959 0 0 0
14 1 4 0.8948539686002976 -2.2639886108037617 -5.312362970992371 0 0 0
6 1 4 0.3692348783202371 -1.465350982028403 -3.15899550958555 0 0 0
17 1 1 -7.911003456940725 6.394604669084831 -3.2847323355216633 0 0 0
19 1 2 -4.724632169402476 5.488478652714059 -2.3629625111176513 0 0 0
2 1 1 0.07043375538628166 0.17639628002391447 -0.17354486996190804 0 0 0
3 1 2 -1.1293341672709265 -1.6663793305227887 -1.560909643935161 0 0 0
4 1 2 -1.4993217540922381 1.4528569289242272 1.4594162096096932 0 0 0
5 1 2 2.3011478774449836 -0.606697964449504 0.9084551046431302 0 0 0
8 1 3 3.0510214685015975 1.4615557135502018 0.9044133017667044 0 0 0
21 1 2 -8.406014297253742 5.390811308505313 -0.03341735447122618 0 0 0
22 1 4 -4.0148940442848255 4.249795340043443 -0.6890642286764004 0 0 0
7 1 4 -2.4123352839762604 2.528061155887325 -0.22887943389097928 0 0 0
18 1 1 -6.3265005793693465 6.769365992689172 -0.7650290786598049 0 0 0
20 1 2 -6.888384240428111 9.182942100254966 -1.5517839067654662 0 0 0
23 1 3 -4.872948860534022 9.857565021997363 -0.9835373421257253 0 0 0
1 1 1 -0.7508467743136755 -1.8804418013849726 1.8500448413540707 0 0 0
24 1 3 -8.554734098641468 6.564245916320649 1.8215580246512046 0 0 0
Velocities
12 0.5234257128414513 -0.413448467353749 0.10839942821823344
9 0.42137988602815346 -0.1307542618819911 0.3298156816434833
15 0.5495031603170716 -0.3651359036788513 -0.21216524326899397
16 0.35173580315590935 -0.01222490378694378 0.6438518275497017
13 0.32672213731770827 0.10265106262408179 0.5983141356113949
10 0.40401785722221645 0.004774789092464965 0.17603452973723085
11 0.3732962542477241 0.23620658187389376 -0.07772031101939694
14 0.293973912040178 0.5231446713805445 -0.05274653363550849
6 0 0 0
17 0.38572582494913077 -0.20018101701534013 0.04866848067881464
19 0.41682630626358447 -0.0751987519085593 0.06402155574650642
2 0 0 0
3 0 0 0
4 0 0 0
5 0 0 0
8 0 0 0
21 0.42590424174125374 -0.13143575826944678 0.07600966007154268
22 0.46040343327543454 -0.009218300404028837 0.09437019541632069
7 0 0 0
18 0.38031089525633704 -0.08449404912420623 0.034867240617758345
20 0.3017546565764182 -0.12274523016092512 -0.02637505231348842
23 0.2818207544419273 -0.044075863407094594 -0.0490705989870885
1 0 0 0
24 0.39345616339259915 -0.0879747683347712 0.04591529759375202
Bonds
1 3 12 15
2 1 9 10
3 3 13 16
4 2 10 11
5 2 10 12
6 2 10 13
7 3 11 14
8 4 14 6
9 3 6 3
10 1 17 18
11 2 2 3
12 2 2 4
13 2 2 5
14 3 4 7
15 3 5 8
16 3 21 24
17 3 22 19
18 4 7 22
19 2 18 19
20 2 18 20
21 2 18 21
22 3 20 23
23 1 1 2
Angles
1 1 11 14 6
2 1 3 6 14
3 1 19 22 7
4 1 4 7 22
Dihedrals
1 2 10 11 14 6
2 1 11 14 6 3
3 2 18 19 22 7
4 2 2 3 6 14
5 2 2 4 7 22
6 1 4 7 22 19
Out/I213.3.1000000.dat (output of write_data)
LAMMPS data file via write_data, version 22 Jul 2025, timestep = 910000, units = nano
24 atoms
4 atom types
22 bonds
4 bond types
2 angles
1 angle types
2 dihedrals
2 dihedral types
-20 20 xlo xhi
-20 20 ylo yhi
-20 20 zlo zhi
Masses
1 9.42478
2 9.42478
3 9.42478
4 9.42478
Bond Coeffs # harmonic
1 828 3
2 828 2.6
3 828 2.2
4 828 2.3439
Angle Coeffs # cosine/shift/exp
1 828 138.546 20
Dihedral Coeffs # cosine/shift/exp
1 828 169.821 200
2 828 -176.209 200
Atoms # molecular
21 1 2 -8.809663223102525 1.6481866190331949 -1.674266794589877 0 0 0
6 1 3 0.3692348783202371 -1.465350982028403 -3.15899550958555 0 0 0
17 1 1 -8.45315779605319 3.9771685236016143 -4.178749399331056 0 0 0
19 1 2 -5.57938583562913 4.4909726179063005 -2.3618492862455502 0 0 0
20 1 2 -9.621833020736219 5.928815907935563 -1.6001923114990941 0 0 0
24 1 3 -9.951606364989928 1.727839794191178 0.20446183075161306 0 0 0
2 1 1 0.07043375538628166 0.17639628002391447 -0.17354486996190804 0 0 0
3 1 2 -1.1293341672709265 -1.6663793305227887 -1.560909643935161 0 0 0
4 1 2 -1.4993217540922381 1.4528569289242272 1.4594162096096932 0 0 0
5 1 2 2.3011478774449836 -0.606697964449504 0.9084551046431302 0 0 0
8 1 3 3.0510214685015975 1.4615557135502018 0.9044133017667044 0 0 0
18 1 1 -7.877805477850359 4.035390822158192 -1.2350137122777267 0 0 0
22 1 4 -4.57675491069403 3.216039638340589 -0.8754855550971391 0 0 0
7 1 4 -2.4123352839762604 2.528061155887325 -0.22887943389097928 0 0 0
23 1 3 -8.54697904358565 7.218767081435103 -0.17867378784746402 0 0 0
1 1 1 -0.7508467743136755 -1.8804418013849726 1.8500448413540707 0 0 0
15 2 3 -6.931255591750357 6.795619221925736 5.756022003117857 0 0 0
12 2 2 -7.355811698766184 8.53724814645925 4.480686432956907 0 0 0
11 2 2 -10.331964451266497 9.271486696423734 7.577830676251562 0 0 0
9 2 1 -10.023431718820403 10.70688870943151 4.468471487795439 0 0 0
10 2 1 -8.0278685844727 9.931301088033726 6.569945146852064 0 0 0
16 2 3 -5.523882429174659 12.257121635978878 6.435246156566482 0 0 0
13 2 2 -7.705038767306363 12.494000441553919 6.272625507843174 0 0 0
14 2 3 -9.692825718746114 9.988863488923599 9.556939625619634 0 0 0
Velocities
21 -0.1518415086613703 0.3233771603609142 -0.1494749561526549
6 0 0 0
17 -0.3581859526214173 0.2712178992657632 -0.22735167567559064
19 -0.03724349305442411 0.06560582839507603 -0.676837649879446
20 0.19230730628563147 0.38722250422350796 -0.06565358833412346
24 0.1449212331837823 0.4304716398628484 0.026365108254468028
2 0 0 0
3 0 0 0
4 0 0 0
5 0 0 0
8 0 0 0
18 0.10162964234267624 0.25524129366584064 -0.3169065140962693
22 0.09355549135796579 0.0021924928542482736 -0.8194614606632595
7 0 0 0
23 0.5124208347711648 0.3175628749898772 -0.2444889496235933
1 0 0 0
15 0.011689226201220537 0.3476004241473308 -0.31804507511314745
12 -0.32252005984924753 0.2820180722684424 -0.29634845416857003
11 0.07039411826727829 0.10326493057625882 0.12359325925708511
9 -0.5073304208697133 0.05671024359297183 0.04477585437774159
10 -0.13328250447038548 0.2740965025141501 -0.230190304952672
16 -0.34925529919946596 0.4821719156484023 -0.652218832503613
13 -0.39311945048643615 0.2943880741447474 -0.3374214195270685
14 0.3005348742779862 0.2033759317399952 0.012983290019840527
Bonds
1 3 21 24
2 1 17 18
3 3 20 23
4 2 2 3
5 2 2 5
6 2 2 4
7 3 3 6
8 3 4 7
9 3 5 8
10 2 18 19
11 2 18 20
12 2 18 21
13 3 22 19
14 4 7 22
15 1 1 2
16 3 12 15
17 2 12 10
18 2 11 10
19 1 10 9
20 3 13 16
21 2 13 10
22 3 14 11
Angles
1 1 19 22 7
2 1 4 7 22
Dihedrals
1 2 18 19 22 7
2 1 4 7 22 19
rxn1_pre.txt
molecule template:
12 atoms
10 bonds
0 angles
0 dihedrals
0 impropers
Types
1 1 # helix top
2 1 # helix bottom
3 2 # target arm
4 2 # side arm, edge
5 2 # side arm, edge
6 3 # target arm, initiator
7 3 # target arm, initiator
8 2 # side arm, edge
9 2 # side arm, edge
10 2 # target arm
11 1 # helix bottom
12 1 # helix top
Coords
1 0.0 0 0
2 0.0 0 0
3 0.0 0 0
4 0.0 0 0
5 0.0 0 0
6 0.0 0 0
7 0.0 0 0
8 0.0 0 0
9 0.0 0 0
10 0.0 0 0
11 0.0 0 0
12 0.0 0 0
Bonds
1 1 1 2
2 2 2 3
3 2 2 4
4 2 2 5
5 3 3 6
6 3 7 10
7 2 10 11
8 2 9 11
9 2 8 11
10 1 11 12
rxn1_post.txt
molecule template:
12 atoms
11 bonds
2 angles
3 dihedrals
0 impropers
Types
1 1 # helix top
2 1 # helix bottom
3 2 # target arm
4 2 # side arm, edge
5 2 # side arm, edge
6 4 # target arm, initiator
7 4 # target arm, initiator
8 2 # side arm, edge
9 2 # side arm, edge
10 2 # target arm
11 1 # helix bottom
12 1 # helix top
Coords
1 0.0 0 0
2 0.0 0 0
3 0.0 0 0
4 0.0 0 0
5 0.0 0 0
6 0.0 0 0
7 0.0 0 0
8 0.0 0 0
9 0.0 0 0
10 0.0 0 0
11 0.0 0 0
12 0.0 0 0
Bonds
1 1 1 2
2 2 2 3
3 2 2 4
4 2 2 5
5 3 3 6
6 3 7 10
7 2 10 11
8 2 9 11
9 2 8 11
10 1 11 12
11 4 6 7
Angles
1 1 3 6 7
2 1 10 7 6
Dihedrals
1 1 3 6 7 10
2 2 2 3 6 7
2 2 11 10 7 6
rxn1_map.txt
map file:
12 equivalences
4 edgeIDs
5 constraints
InitiatorIDs
6
7
EdgeIDs
4
5
8
9
Equivalences
1 1
2 2
3 3
4 4
5 5
6 6
7 7
8 8
9 9
10 10
11 11
12 12
Constraints
angle 3 6 7 108.5463 168.5464 # 138.5464 +- 30
angle 10 7 6 108.5463 168.5463 # 138.5464 +- 30
dihedral 3 6 7 10 139.820904 -160.179096 # 169.820904 +- 30
dihedral 2 3 6 7 93.7912138 -86.2087862 # -176.2087862 +- 90
dihedral 11 10 7 6 93.7912138 -86.2087862 # -176.2087862 +- 90