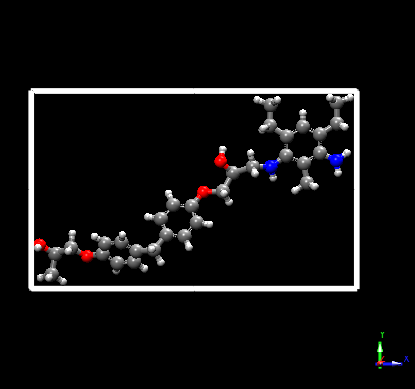
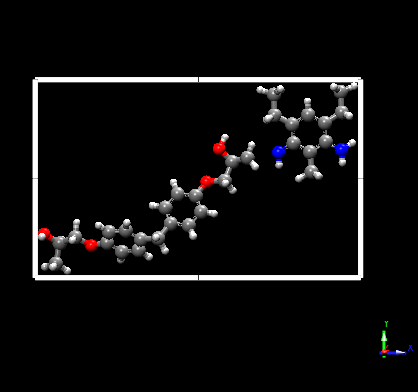
Hello, I am trying to simulate the bond breaking in a crosslinked epoxy. I tried bond/react and I have come up with some issues.
Firs of all i present the pre and the post state of my reaction
Pre state:
Post State:
You can notice that the bond between the Nitrogen and Carbon atom is broken.
I tried to generate the molecule files by my self based on the individual data files of the two geometries. I have an issue on the atom types compatability. In fact when a bond is broken automatically we need a new atom type to describe the atom conected only to 3 atoms instead of four. So when the simulation initiates I have 11 bond types and 6 atom types (and so on with the angles dihedrals etc) but as the simulation runs and the bond is broken I have less bond types and more atom types.
In the begining of the simulation should I include all the parameters for the force field for all the bonds that i have and i will have? Will this be a problem?
Or if you have any advice on how i can simulate that it would be very helpful
Thank you in advance,
For questions about fix bond/react you need to contact the developer. He does follow this forum and responds to answers here, but he may not notice your post instantly. Thus you can trigger an email notification (in case he is idle or not logged in) by adding @jrgissing as a mention to your post like I just did.
Yes, all atom, bond, etc. types that may appear during the simulation need to be specified in the initial data file. You mentioned a potential issue regarding atom types compatibility. I highly recommend using the relatively new addition to LAMMPS called ‘type labels’, which enables replacing numeric types with character strings instead. For example, you could have an atom type ‘c1’, which could match the atom type in your force field. Using type labels removes the need to make sure that different files are using ‘compatible numbers’ for atom, bond, etc. types.
Dear sir, @jrgissing
Thank you for your reply!
You are correct about that but my software that creates the forcefield is still producing them by numbers. I have another short question for you. I implemented the fix bond/react to a simple smaller molecule of polyethylene to break it in half. I created also two new hydrogen atoms to be placed in the broken area of the C-C bond. It seems in the visualisation of trajectory that the ney atoms created do not participate in the integration algorithm (they do not move at all) Is this a visualisation issue?
It is hard to say. Can you provide a short example that reproduces the problem? If you cannot attach files, pasting links work well (e.g., to files on Google Drive).
@jrgissing Hello again,
I tryied to simulate a more simple reaction to see how the command works on a crosslinking prosedure. I managed to generate the pre and post tempates with moltemplate so as to be compatible with the bond names etc. I read that there may be some issues with the update of the bond types on pre and post reaction templates so i fixed that but when i run a simulation with two edge atoms i got
Fix bond/react: Excessive iteration of superimpose algorithm. Please check that all pre-reaction template atoms are linked to an initiator atom, via at least one path that does not involve edge atoms. (src/REACTION/fix bond react.cpp:1428)
The error message is trying to tell you the issue. Specially, atoms 42 and 43 are ‘past’ the edge atom (atom 22), when tracing from the initiator atom. They should be deleted. Note also that your templates have many more atoms than necessary, which could affect efficiency or parallel issues.
Regarding AutoMapper, I know that it was designed specifically to decrease the complications involved with bond types compatibility. So, if you have issues that software, you should contact its author Matt Bone ([email protected])
I fixed the problem with the help of automapper. In fact what i did wrong is that I used the cleanup tool of moltemplate and then also the clear module of automapper and there was an incompatibility of the bond types between the pre and post geometries and the autromapper tool could not work in a right way. Thanks a lot for your responses and your help!
@jrgissing Hello again,
I want to ask about an error im facing while I try to have two different reaction templates in the same input file as the example in the examples file of the lammps documentation.
In fact my code runs normally if only one reaction template is preset and when I have two templates it pops up a weird error like
You must confirm that this issue still exists with the 15 June 2023 version. This may be a bug that has already been fixed in the feature releases and nobody likes to track down and debug the same bug multiple times.
[DESKTOP-7KSVN7D:23509] *** Process received signal ***
[DESKTOP-7KSVN7D:23509] Signal: Segmentation fault (11)
[DESKTOP-7KSVN7D:23509] Signal code: (128)
[DESKTOP-7KSVN7D:23509] Failing at address: (nil)
[DESKTOP-7KSVN7D:23509] [ 0] /lib/x86_64-linux-gnu/libpthread.so.0(+0x14420)[0x7fad87021420]
[DESKTOP-7KSVN7D:23509] [ 1] lmp(+0x2676f4)[0x7fad8732c6f4]
[DESKTOP-7KSVN7D:23509] [ 2] lmp(+0x273ce1)[0x7fad87338ce1]
[DESKTOP-7KSVN7D:23509] [ 3] lmp(+0x275dbb)[0x7fad8733adbb]
[DESKTOP-7KSVN7D:23509] [ 4] lmp(+0x151956)[0x7fad87216956]
[DESKTOP-7KSVN7D:23509] [ 5] lmp(+0x15bd98)[0x7fad87220d98]
[DESKTOP-7KSVN7D:23509] [ 6] lmp(+0x100be0)[0x7fad871c5be0]
[DESKTOP-7KSVN7D:23509] [ 7] lmp(+0x101315)[0x7fad871c6315]
[DESKTOP-7KSVN7D:23509] [ 8] lmp(+0xdc7de)[0x7fad871a17de]
[DESKTOP-7KSVN7D:23509] [ 9] /lib/x86_64-linux-gnu/libc.so.6(__libc_start_main+0xf3)[0x7fad86694083]
[DESKTOP-7KSVN7D:23509] [10] lmp(+0xdda0e)[0x7fad871a2a0e]
[DESKTOP-7KSVN7D:23509] *** End of error message ***
[DESKTOP-7KSVN7D:23510] *** Process received signal ***
[DESKTOP-7KSVN7D:23510] Signal: Segmentation fault (11)
[DESKTOP-7KSVN7D:23510] Signal code: (128)
[DESKTOP-7KSVN7D:23510] Failing at address: (nil)
[DESKTOP-7KSVN7D:23510] [ 0] /lib/x86_64-linux-gnu/libpthread.so.0(+0x14420)[0x7f6b0bac1420]
[DESKTOP-7KSVN7D:23510] [ 1] lmp(+0x2676f4)[0x7f6b0bdcb6f4]
[DESKTOP-7KSVN7D:23510] [ 2] lmp(+0x273ce1)[0x7f6b0bdd7ce1]
[DESKTOP-7KSVN7D:23510] [ 3] lmp(+0x275dbb)[0x7f6b0bdd9dbb]
[DESKTOP-7KSVN7D:23510] [ 4] lmp(+0x151956)[0x7f6b0bcb5956]
[DESKTOP-7KSVN7D:23510] [ 5] lmp(+0x15bd98)[0x7f6b0bcbfd98]
[DESKTOP-7KSVN7D:23510] [ 6] lmp(+0x100be0)[0x7f6b0bc64be0]
[DESKTOP-7KSVN7D:23510] [ 7] lmp(+0x101315)[0x7f6b0bc65315]
[DESKTOP-7KSVN7D:23510] [ 8] lmp(+0xdc7de)[0x7f6b0bc407de]
[DESKTOP-7KSVN7D:23510] [ 9] /lib/x86_64-linux-gnu/libc.so.6(__libc_start_main+0xf3)[0x7f6b0b134083]
[DESKTOP-7KSVN7D:23510] [10] lmp(+0xdda0e)[0x7f6b0bc41a0e]
[DESKTOP-7KSVN7D:23510] *** End of error message ***
Primary job terminated normally, but 1 process returned
a non-zero exit code. Per user-direction, the job has been aborted.
mpirun noticed that process rank 0 with PID 0 on node DESKTOP-7KSVN7D exited on signal 11 (Segmentation fault).
[DESKTOP-7KSVN7D:23505] 3 more processes have sent help message help-btl-vader.txt / cma-permission-denied
[DESKTOP-7KSVN7D:23505] Set MCA parameter “orte_base_help_aggregate” to 0 to see all help / error messages
looks you found a tricky memory-related bug that is triggered when creating new atoms. I am traveling the next couple days, but will take a closer look soon.
You can avoid the error by not deleting and recreating the hydrogen atom. Indeed, I would not recommend this approach in your case, even without the bug. You can simply reconnect the hydrogen atom where it needs to be by adjusting its bond connectivity.
@jrgissing hello again, I tried a different aproach that does not contain atom creation. The two reactions seperately work perfect but together an error pops up
fix fxrct all bond/react stabilization yes statted_grp .03 react rxn2 all 1 2.0 5.0 mol3 mol4 r2_automap.data stabilize_steps 100
ERROR: Bond/react: A deleted atom cannot remain bonded to an atom that is not deleted (src/REACTION/fix_bond_react.cpp:2477)
Last command: fix fxrct all bond/react stabilization yes statted_grp .03 react rxn2 all 1 2.0 5.0 mol3 mol4 r2_automap.data stabilize_steps 100
The error is obvious but i checked the map files and ther is no deleted atom involved in another bond in the reaction 2. Attached you will find my files…
Thanks a lot! Have a nice day! run.zip (77.4 KB)
ok, the issue is that you listed the same molecule for both your pre- and post-reaction templates, for your second reaction. This typo is probably what caused the memory issue mentioned above as well. If you have time, do you mind confirming this?