I Need to apply fix command based on MoleculeID and atomID (need odd atoms) because of 32 group limit. I cannot delete groups because they would rely on fixes. Can I apply a fix to atoms in a group that only have a certain molecule ID? i.e. have 1 big group and choose atoms from it to apply fix based on molecule ID instead of having a group for each molecule?
something like
fix (fix name) (big group id) (style) (args) Condition that applies fix based on MoleculeID && odd atomID
If you need context on the current simulation (not required to read this):
I am running a simulation of multiple Hexamer rings that composed of 3 two state dumbbells.
These dumbbells have a harmonic bond and each monomer in the dumbbell undergoes langevin dynamics. These dumbbells have the chance to randomly switch states every timestep, which changes the equilibrium spring length of the bond and the dampening factor of one of the monomers. This gives the dumbbell a self propelled motion.
Anyway, I am able to run one of these hexamers just fine, but running more than 32 is a nightmare because of the 32 group limit. I loop of each hexamer moleculeID, update the states for each one, then run 1 timestep, repeat. If i make a group for each hexamer, i cannot delete it because it has its own fix for langevin dynamics which is updated after every timestep because the damping factor may change. Therefore the only option I see is to define the fix based on The moleculeID and atomID instead of the group, but I cannot do this. Or is there a way to update fix on certain atoms of a certain molecule within a group?
cant attach files, I am a new member, I can always email.
Yes, fix rigid/small does that, for example. The fix is applied to the group of atoms defined by the fix group and then rigid bodies are defined by molecule IDs, i.e. all atoms with the same molecule ID are one rigid object.
A more general way to do this kind of thing would be to use the “chunk” mechanism: 8.3.2. Use chunks to calculate system properties — LAMMPS documentation
You use compute chunk/atom to group atoms into chunks, and then write a fix that treats all atoms in the same chunk as one item. No more limitations to 32 groups.
Even though you cannot attach files, you can paste script text into your posts. If you turn on “verbatim mode” with three backticks
```
like this keep scrolling I'm sorry, the princess is in another castle
```
the forum formatting will not just skip special symbols but add horizontal or vertical scrollbars as needed.
Can you track your particle statuses by changing particle type? For example
set atom SOME_ID type NEW_TYPE
Since fix langevin allows you to scale damping factors based on particle type (with the scale keyword), changing types automatically changes their damping factor.
The only element left would be to dynamically change bond types – presumably it would be easy to write a fix that looks like
that, every 100 steps, checks the types of all bonded atoms and changes bonds between atom-types 1 and 1 to bond-type 5, between atom-types 1 and 2 to bond-type 1, between atom-types 1 and 3 to bond-type 3 …
Thank you! It seems like chunks can do what I am looking for if I understand it correctly - Make a group of all atoms, then separate that group into chunks based on MoleculeID and odd atomID. So chunk 1 would be the odd numbered atoms of molecule 1 and so on. then apply a separate fix for langevin motion for each chunk
thanks for your suggestion, so it seems that I can make the odd numbered atoms atomID in the Molecule the same as the moleculeID to match the looping. Then make that a type. then apply fix langevin in the loop based on the current type. Axel suggested I could use chunks, so I will try that as well.
Here is my current loop over moleculeID (it runs with no errors). There are 5 molecules now for simplicity, but I would get a group error if there were to be more than 32 molecules
#Simulation loop for running 1 timestep after each molecule is updated independently
label loop
#inner loop over molecules
variable mol_id loop 5
label mol_loop
variable bond_type equal "v_mol_id" # Set the bond type dynamically to the molecule ID
#Create a condition combining molecule ID and odd atom ID to select active monomers in a certain molecule
variable condition atom "mol == v_mol_id && id % 2 == 1"
#Group atoms that satisfy the condition
group odd_atoms${mol_id} variable condition
#initial langevin dynamics for active monomer
fix langevin${mol_id} odd_atoms${mol_id} brownian 0.0001 12908410 gamma_t 10 rng gaussian
#Print current state
print "Current step: ${current_step}, Current state: ${current_state}"
#Generate a random number
variable rand equal random(0,1,12345*${mol_id})
#Transition logic for state A (0)
if "${current_state} == 0" then "variable next_state equal 0"
if "${current_state} == 0 && ${rand} < ${p_AtoB}" then "variable next_state equal 1"
#Transition logic for state B (1)
if "${current_state} == 1" then "variable next_state equal 1"
if "${current_state} == 1 && ${rand} < ${p_BtoA}" then "variable next_state equal 0"
#Update the current state
variable current_state equal ${next_state}
#Update bond length (equllibrium spring lengths in molecule) and damping based on the new state
if "${current_state} == 0" then "bond_coeff ${bond_type} 500.0 0.5"
if "${current_state} == 1" then "bond_coeff ${bond_type} 500.0 1.5"
#update dynamics of odd atoms group
unfix langevin${mol_id}
if "${current_state} == 0" then "fix langevin${mol_id} odd_atoms${mol_id} brownian 0.0001 12908410 gamma_t 10 rng gaussian"
if "${current_state} == 1" then "fix langevin${mol_id} odd_atoms${mol_id} brownian 0.0001 12908410 gamma_t 50 rng gaussian "
#end molecule loop
next mol_id
jump SELF mol_loop
#Run one timestep
run 1
#Increment step counter
variable current_step equal ${current_step}+1
#Check if total simulation time is reached
if "${current_step} >= ${total_steps}" then "jump SELF end"
#Loop back
jump SELF loop
I have tried the scale keyword with fix Langevin, it does not work, for the damping ceoff must be updated for each molecule at every timestep. Therefore, when I loop over my molecules I apply the fix each time. This means the fix is overwritten at each iteration of the molecule loop, so only the last molecule changes damping factors. My loop is below
I have added all the atoms I want to change the damping factor of to a group called active. Then set the Atom IDs of the atoms in this group equal to the moleculeID for indexing.
# Simulation loop
label loop
# Inner loop over molecules
variable mol_id loop 5
label mol_loop
variable bond_type equal "v_mol_id"
# Initialize langevin dynamics for active monomer, so we can use the unfix command
fix langevin${mol_id} active langevin .0001 .0001 10 12345 scale ${mol_id} 1
# Get current molecule's state
variable current_mol_state equal v_state_${mol_id}
# Generate a random number
variable rand equal random(0,1,${mol_id})
# Transition logic for current molecule
if "${current_mol_state} == 0 && ${rand} < ${p_AtoB}" then "variable state_${mol_id} equal 1"
if "${current_mol_state} == 1 && ${rand} < ${p_BtoA}" then "variable state_${mol_id} equal 0"
print "Current bond : ${bond_type}"
# Print current state
print "Current step: ${current_step}, Current state of molecule ${mol_id}: ${current_mol_state}"
# Update bond length based on new state
if "${current_mol_state} == 0" then "bond_coeff ${bond_type} 5000 1"
if "${current_mol_state} == 1" then "bond_coeff ${bond_type} 5000 2.5"
# Update dynamics of odd atoms group
unfix langevin${mol_id}
if "${current_mol_state} == 0" then "fix langevin${mol_id} active langevin .0001 .0001 1000 12345 scale ${mol_id} 1"
if "${current_mol_state} == 1" then "fix langevin${mol_id} active langevin .0001 .0001 1000 12345 scale ${mol_id} 10"
# End molecule loop
next mol_id
jump SELF mol_loop
# Run one timestep
run 1
# Increment step counter
variable current_step equal ${current_step}+1
# Check if total simulation time is reached
if "${current_step} >= ${total_steps}" then "jump SELF end"
# Loop back
jump SELF loop
I have played around with chunks for the past week. I do not see any documentation on applying a fix like fix brownian or fix langevin (these are the ones i need) to a chunk of a group instead of the group itself. Is applying these a fix to a chunk possible? This seems like the only way to get around my issue
So there is no way to use fix brownian or fix langevin on only specific atoms within a group? (while not applying anything to any other atoms within the group)
something like fix (ID) (groupID) (brownian or langevin) … (condition to apply to only atoms of certain atom or moleculeID in group ID while ignoring all others in groupID)
Please describe exactly what you are trying to do. Draw (or digitally render) the monomers, dumbbells, and hexamers, and how they are meant to be changing state and what exactly happens when they do. (This is work you would have to do for a publication anyway.)
In your initial post you said:
When you said “one of the monomers” I assumed this was one particle in each dumbbell. But your LAMMPS script attempts to change the scaling for both particles in the dumbbell. Only one of these can be true, and I can’t help you more specifically until I know which.
I can appreciate your desire to only communicate necessary information to try and be helpful – but if we do not have enough information we cannot give you the help you are looking for at all.
Got it, Sorry i think I switched from a hexamer configuration to a dumbbell configuration without saying because the dumbbells are an easier case, so I will give you the dumbbell code.
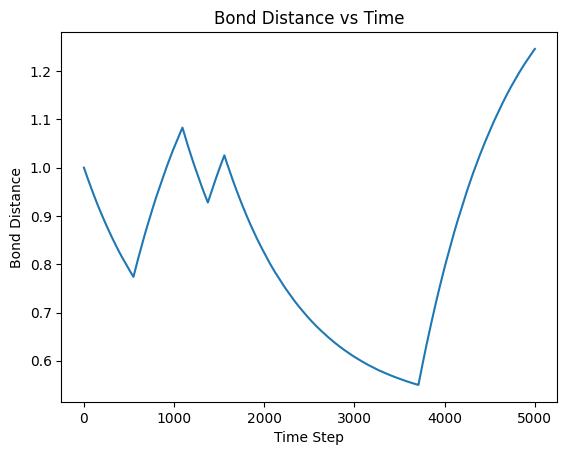
The Dumbbell is composed of two atoms with a harmonic bond. These atoms follow overdamped brownian motion. The dumbbell has two states, A and B. States are determined via a random number generator every timestep. In state A, the dumbbell’s harmonic potential has equillibrium length, say 1, and state B has eq length, say 2. This can be visualized with a graph of the distance between the two dumbbell atoms
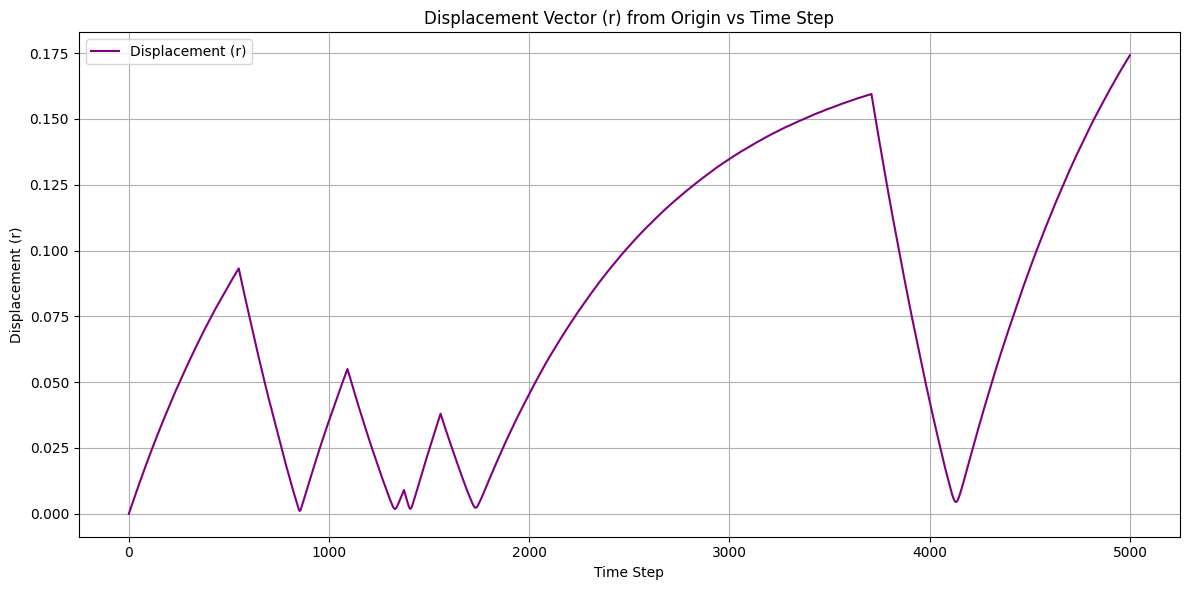
in state B, the damping factor of the 2nd atom in the dumbell will change. This causes self-propelled motion, as shown in the attached COM displacement vector graph.
Imagine a dumbbell growing and contracting with different velocities on each side. Its COM will move
I have successfully made a codes for both a single one and up to 32 dumbbells(both .in filesAttached to this message), But cannot make a code for simulating over 32 independent dumbbells because I need to changed the damping coefficients independently with fix brownian or langevin(change states and damping independently). I need to simulate more than 32 Dumbbells
Single Dumbbell dumbell_input.dat (236 Bytes)
These show two methods to achieve something like what you want (using fix halt on a global “reporter variable” to trigger molecular reassignments, consisting of a type reassignment to enable fix langevin per-type drag scaling and a combination of delete_bonds and create_bonds to replace one bond type by another). Neither chunking nor using fancy group-selectors to influence the integrator is required.
One uses independent per-atom random variables to toggle independent molecules; the other, which is preferable in my opinion, uses a single global random variable to either not change any molecule types or change a single molecule type at the specified probability.
Since these scripts are self-contained and fully operational, you should be able to learn what you need to know by carefully checking and reverse-engineering them as needed. I will note that, from a theoretical perspective, you are very aggressively “snapping” the bonds from one length to another. Please double-check the amount of power (dissipated bond energy per time) generated during a typical bond length change and reconsider the physicality of your model.
You could change int igroup, groupbit to tagint in fix.h to get 64 groups when compiling with BIGBIG but you would need to keep your own separate fork on GitHub and sync your fork periodically to avoid conflicts.
You can’t contribute that change back into develop unless you get permission from @akohlmey
ps: you can go up to 1024 groups with uint1024_t by linking boost multiprecision library but then you definitely need to keep your own fork.
I’m assuming that you mean changing your input script rather than the LAMMPS source code?
(It’s not a frequent distinction since most people don’t need highly modified functionalities – but when those are needed, it’s really helpful to make a distinction between the two! “Code” is ambiguous in that context so I tend to at least distinguish between “code” and “script”.)