Hi everyone,
I am using fix deposit to insert atoms (interacting with a MEAM potential) into a box as a gas, with a velocity range of some -v to +v velocity units.
What I want to achieve is having some ‘hot’ atoms being inserted, which then gradually cool down. For this purpose, I have coupled these atoms to a thermostat.
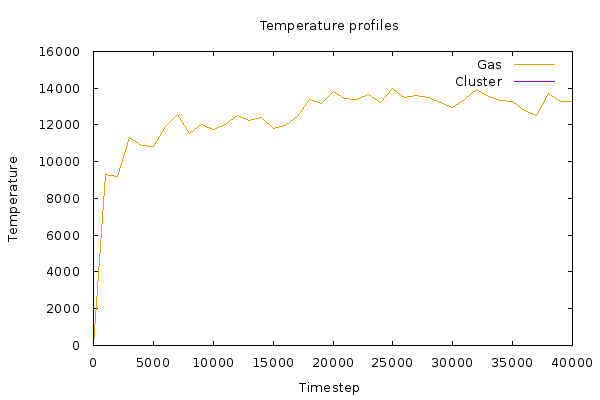
I have noticed that the temperature of this group of atoms is shooting up to unphysical amounts (pls see attached graph).
(This behaviour was noticed before but the story was incomplete).
I’ve stripped down my code to the bare minimum and noticed the problem is in the following lines (being run on 16 Mar 2018 version of LAMMPS):
units metal
atom_style atomic
boundary p p p
lattice sc 1.0 origin 0 0 0
pair_style eam/fs and pair_coeff definitions (for this, I have compiled lammps with MEAM and USER-MEAMC packages)
region sbox block -15 15 -15 15 -15 15 side in
create_box 2 sbox
neigh_modify every 1 delay 0 check yes
group gas empty
compute tg gas temp
compute_modify tg dynamic/dof yes extra/dof 0 #to account for dynamic changing of group size
timestep 0.001
fix f1 gas nvt temp 3000 400 0.01
fix f2 gas deposit 100 1 200 156483 near 1.2 attempt 100&
region sbox vx -$v $v vy -$v $v vz -$v $v
fix f3 gas deposit 20 2 800 213543 near 1.2 attempt 100&
region sbox vx -$v $v vy -$v $v vz -$v $v
run 20000
Apart from getting rubbish values, what I have noticed is this—reducing the velocity of deposition brings down the initial temperature, but temp again increases with increasing number of particles.
I even tried to increase run time, but that only equilibrates the system to the higher temperatures of ~14k temperature units.
In this regard, I have some questions:
- I expected the thermostat to adjust the velocities of the atoms accordingly. Is this wrong?
- Am I making a wrong compute of the temperature, and if so, what is the correct method?
- Is there something fundamentally wrong about my model? Are groups associated with fix deposit always going to behave this way?
Sorry for the rather long description of what is perhaps a simple issue, and thanks for reading this far. Any help would be greatly appreciated.
Thanks a lot for your help,
Praneeth