Dear LAMMPS users and developers,
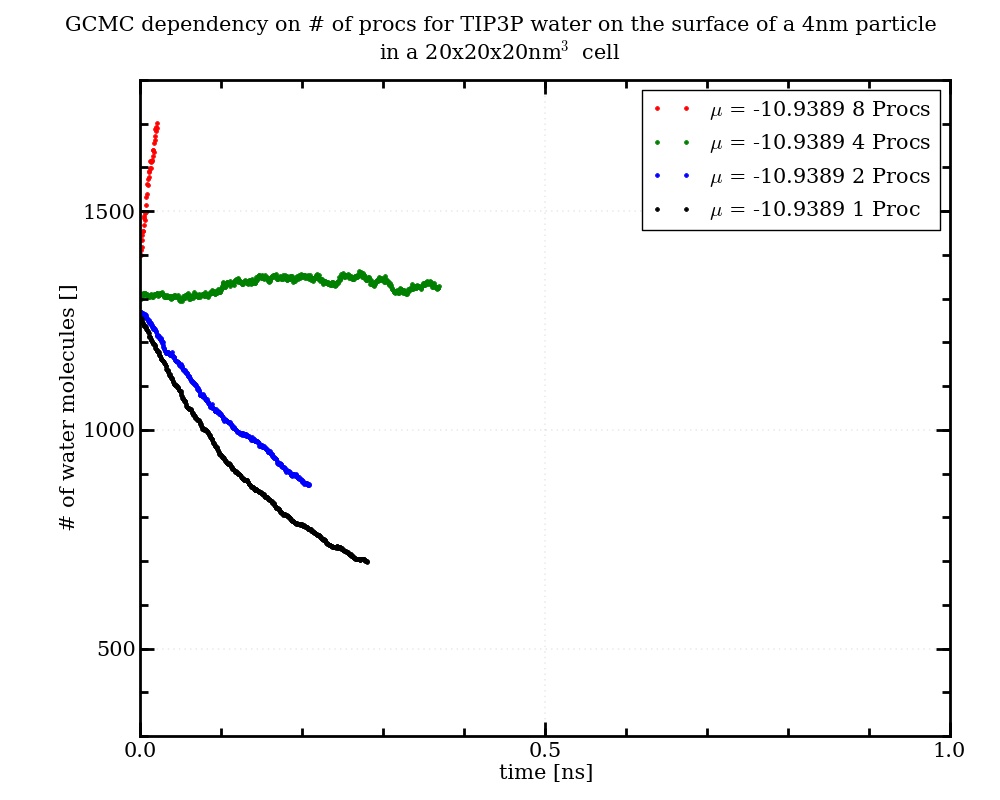
In my research I try to figure out how many water molecules would adsorb on the surface of a TiO2-nanoparticle at different humidities. For this purpose I use the "fix gcmc" to run Grand Canonical Monte Carlo simulations from a datafile containing a nanoparticle with a certain amount of water relaxed on the surface (see attached input script for a simulation at approximately 100% humidity). Curiously, the same simulation running on different numbers of processors produces significantly different numbers of water molecules in the simulation box (see attached diagram and log-files). In the trajectory everything looks ok, while the temperatures in the log-files deviate slightly with varying no. of processors. The no of insertion and deletion attempts is always identical, while the number of accepted attempts deviates. From comparison to gcmc simulations of water on silica and to experimental results on titania the water amount resulting from the simulations on 4 processors seems most likely, which is even more puzzling for me.
Could you help me in figuring out the source of error in my simulations?
I use the latest stable LAMMPS version from the 1st of February 2014 for my simulations on a 4 core Linux machine with multithreading.
Thanks a lot
Jens
in.gcmc_particle_loop (3.14 KB)
gcmc_particle_1proc.log (146 KB)
gcmc_particle_2procs.log (146 KB)
gcmc_particle_4procs.log (147 KB)
gcmc_particle_8procs.log (50.2 KB)
Paul can comment on this.
You should not expect identical answers on different
of procs, due to how random numbers are used
differently. Paul can say whether the differences
you are seeing seem reasonable. Running lots
of independent tests and averaging the results
would be a better test.
Steve
We released a new version of fix gcmc a couple of weeks ago. I’d recommend you download and use this newer version.
Fix gcmc is not well parallelized, so you’ll typically want to run on a single processor unless you’re also doing other heavy parallel calculations. Even so, you should get answers from the same ensemble independent of number of processors. But as Steve notes, the trajectories will diverge when running on different numbers of processors, especially when using this fix. As Steve suggests, it would be best to run lots of independent tests and average the results. Please let me know if you see a dependence on processor count on a test like this with the latest version of the code.
Paul
Dear Paul,
thanks a lot for your help! As the number of molecules differs by a factor of 2 (~700 and ~1300 molecules) between 1 and 4 Processors after 60000 times calling the fix gcmc and as the number of inserted molecules is continously increasing with increasing number of processors, I can not imagine that the statistics is the problem, but I will test this as you suggested. I also tried to use the actual LAMMPS version (28th of June 2014) but my simulations crash only a few steps after invoking the fix gcmc. The oxygen-atoms of the inserted water molecules move very fast directly after insertion while the hydrogen atoms do not move which results in big distances between the bonded atoms. Could it be that I constructed the molecule template file incorrectly? Attached you can find the input and the template that I use. In my data-file the oxygen- and water-atoms are of type 5 and 6 respectively, the OH-bond-type is 3 and the HOH-angle-type is 2. I therefore used these IDs also in the molecule template. Did I understand the manual right, that I do not specify neither the Bond and Angle Coeffs nor the atomic masses in the template file as this is all specified in the data-file?
Best wishes
Jens
tip3p-template (431 Bytes)
in.gcmc_loop_particle (3.14 KB)
There was a bug in the new molecule insertion routine. It is fixed in the attached and in the SVN repo.
Paul
fix_gcmc.cpp (36.5 KB)