I have a problem with “fix rigid”, I’m simulating a lennard-jones fluid with periodic in all directions, after equilibration I define a spherical region inside the field, group them and set its velocity to zero, then I apply “fix rigid” with single keyword on this group. And I also use “neigh_modify exclude” to exclude the interaction of particles inside the rigid body. The problem is that after one time step the position of the rigid body particles change a little bit but then they stay intact as is expected from a rigid body. Any pointer to what is happening, that for the first timestep the constituent of the body are not fixed relative to each other, is appreciated.
It is almost never necessary to us "fix rigid" to immobilize a
molecule. You can accomplish the same thing under NVT conditions by
integrating the equations of motion for the group of mobile atoms.
Here is an example: http://www.moltemplate.org/examples/nanotube+water/run.in.nvt
In this example, I define a group of frozen atoms ("Cgraphene"). The
remaining atoms are "mobile" atoms, and I apply "fix nvt" to these
atoms:
group Cgraphene type 1
group mobile subtract all Cgraphene
fix 1 mobile nvt temp 300.0 300.0 100.0
At high pressures, using "fix rigid" causes LAMMPS to underestimate
the instantaneous pressure: http://lammps.sandia.gov/threads/pngTySiBRzRqL.png
(see green and blue curves, second graph. The correct pressure in
this example should be 10000 atms.)
Steve and Trung may disagree with this approach, but this combination
works well enough for my purposes.
In any case, if you are not running simulations at constant pressure
(or if your immobilized solute is small), then we all agree that you
definitely do not need to use "fix rigid". I hope this helps.
Great that was such a through response…Just a question, by “immobilize” do you mean to have the molecule fixed in space or it still can move as a result of external forces like interaction? Cause what I need just to basically coarse grain a group of particles and look at the effect of solvent on that new generated coarse-grained body.
One small detail: Because the object that I am restraining spans the
entire XY plane, I run NPT so that only the Z dimension of the box can
change. If you have a smaller object you are restraining, you use
"iso" or "aniso" to allow the X and Y dimensions to change also: http://lammps.sandia.gov/doc/fix_nh.html
Great that was such a through response...Just a question, by "immobilize" do
you mean to have the molecule fixed in space or it still can move as a
result of external forces like interaction? Cause what I need just to
this is a constant source of confusion. many people look at
and talk about fix rigid to immobilize atoms and your description
of setting the velocity to zero hints at that.
basically coarse grain a group of particles and look at the effect of
solvent on that new generated coarse-grained body.
then one of the fix rigid styles is the right for you,
or perhaps one of the colloid pair styles may be
a viable alternative.
in any case, it is very difficult to discuss your case
without having an simple and easy to follow example
in hand. there are so many details that can make a
difference, that people may overlook in their description.
in fact, i've seen quite a few cases, where the input
was significantly different from what people claimed
they were doing, which then explained the discrepancies
to what they expected to happen.
Hi,
Here is the script and a movie of the results I get, sorry I couldn’t make a better movie I still have problem in making a movie that can last longer (Axel I’ve also read your post on that in VMD forum) but let me know if the info is not clear. The movie is just 10 frames each taken every 100000 timestep of 0.005 (lj units). I appreciate your help.
Hi,
Here is the script and a movie of the results I get, sorry I couldn't make a
better movie I still have problem in making a movie that can last longer
a movie is a waste of bandwidth in this case. 2-3 images would have done.
(Axel I've also read your post on that in VMD forum) but let me know if the
info is not clear. The movie is just 10 frames each taken every 100000
it is not clear what you mean by "the info is not clear".
if you didn't understand what was written, perhaps you
need to spend a little more effort in trying to understand
and perhaps read up a little on the principles of how these
things work. like many problems in science and elsewhere,
if they seem confusing, it is a sign that you don't yet
understand what you are doing. this is frequently caused
by lack of knowledge of the fundamentals. example: if
you don't understand calculus, linear algebra is impossible
to comprehend, regardless how well it is explained.
timestep of 0.005 (lj units). I appreciate your help.
I think there is a misunderstanding of what I said. What I meant was that if the information I’m providing on my own problem is clear enough or not. Actually by what I’d said in parenthesis I wanted to appreciate the solutions you provided on making a movie in VMD in that forum.
Anyways, I have attached two snapshots of the simulation. In the first one, the rigid body is as it’s formed initially in spherical shape, however, in the second snapshot which is 100000 timestep later its constituents have displaced a bit but they remain intact in later times. I don’t understand why the constituent of the rigid body are displaced if they are rigid? and how I can fix the issue?
The problem is that after one time step the position of the rigid body particles change a little bit but then they stay intact as is >expected from a rigid body. Any pointer to what is happening, that for the first timestep the constituent of the body are not fixed >relative to each other, is appreciated.
I don't understand your Q. You said later that you want the rigid body
to be subject to forces from all the other solvent particles, which
is the only reason to treat it as a rigid body (as opposed to just
not time integrate it like Axel suggested).
In that case, the rigid body will move over time. You can print out
its center of mass position and well as center of mass velocity and forces,
as outputs from the fix rigid command (see its doc page).
Steve,
That’s right, I want it to behave as a rigid body and move over time in the solvent but I want it to retain its initial shape and not deform itself. My question is why the rigid body doesn’t retain its initial shape (rigid0.jpg in previous post) as it moves around, and its constituents are displaced a bit somehow (rigid1.jpg).
I think the only way that could happen is if you are integrating
the atoms in the rigid body more than once. E.g. if they
are also included in a fix nve or fix nvt. If you do this
you should get a warning from LAMMPS. If fix rigid
is the only integrator being used, it should not be possible
for the body atoms to change their relative orientation.
I'll add that images are not very convincing. How are you
sure that all the atoms you think are actually in the rigid
body? Have you printed/examined those coords in a dump file
to insure that have actually displaced differently relative
to each other?
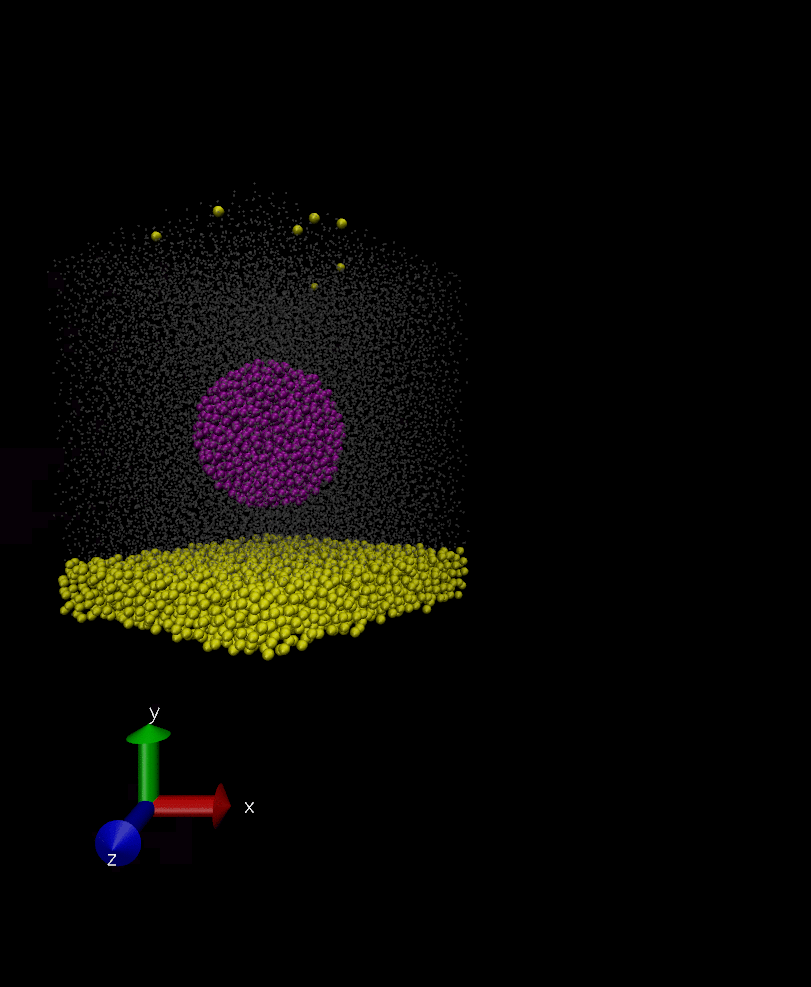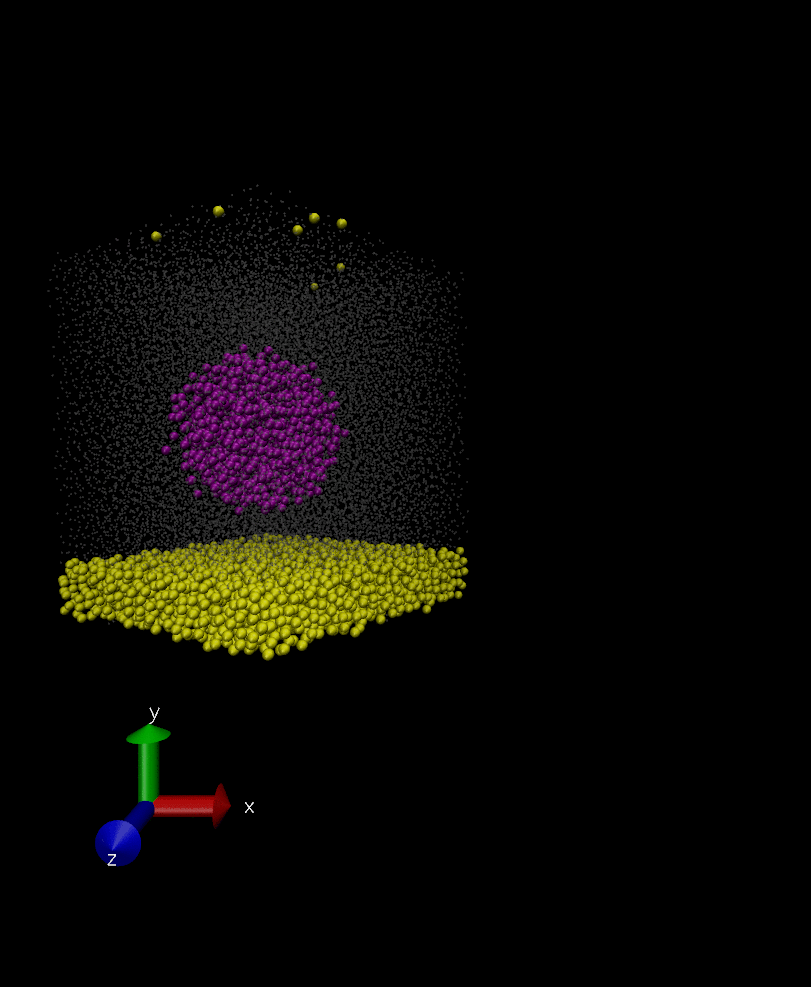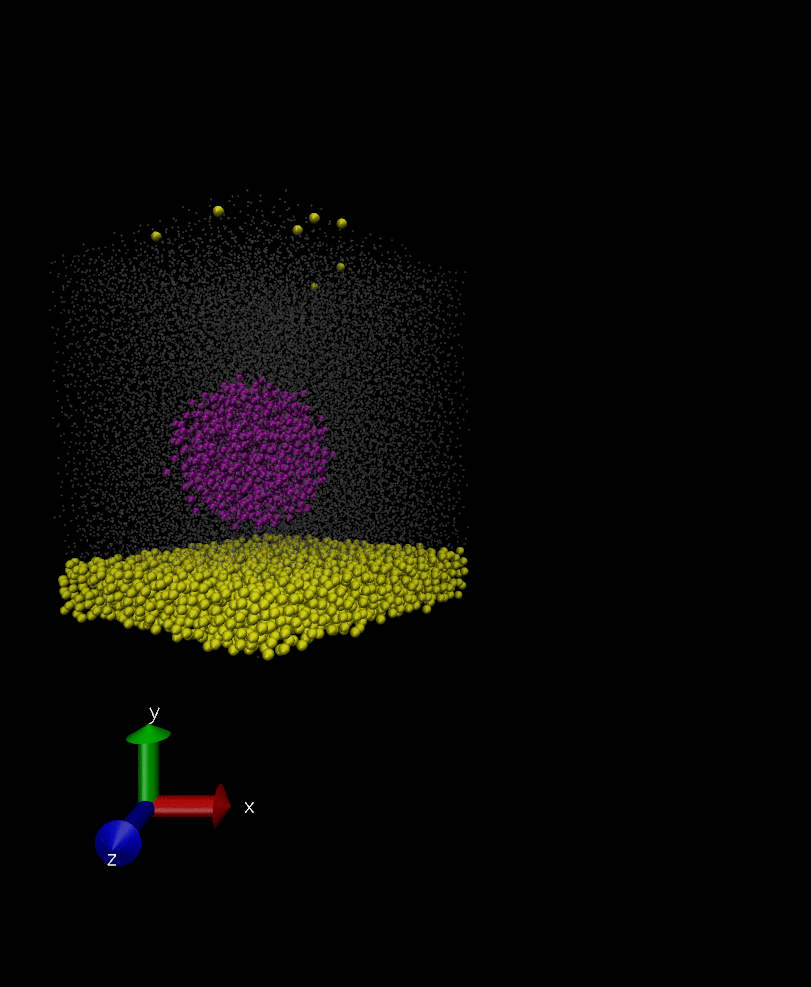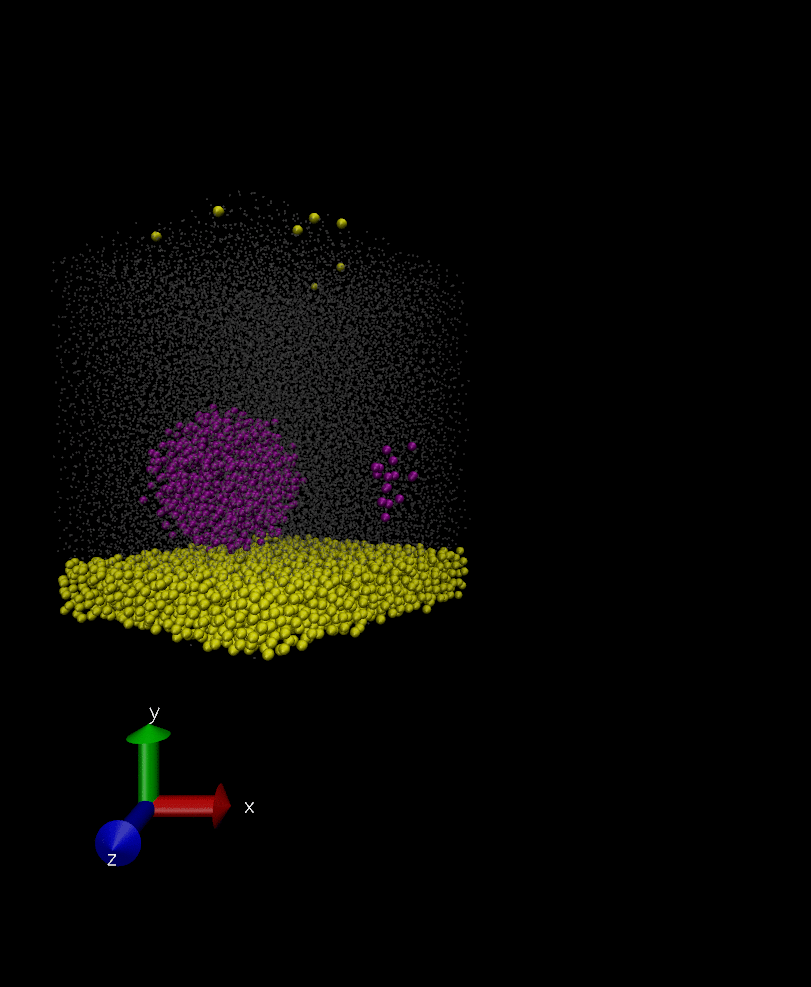
I did a test simulation and this time I’m just including 3 atoms in the rigid body so that I can investigate the problem easier. I wrote out the position of these atoms (rigid body atoms) which form a triangle but the triangle sides are shrinking and expanding over the time. I also tested a case with not using “fix rigid” for the group of three atoms and it turned out that the three atoms remain intact as I’m not including the group in integration, which shows that I’m not double integrating the group when I use “fix rigid”. Any idea what may the problem lie?
I finally could find what the problem was with my simulation. I was creating the rigid body after equilibration and I was not setting the periodic flags of the constituents of the body to be all the same, basically I was not applying following note in the doc page:
IMPORTANT NOTE: To compute the initial center-of-mass position and other properties of each rigid body, the image flags for each atom in the body are used to “unwrap” the atom coordinates. Thus you must insure that these image flags are consistent so that the unwrapping creates a valid rigid body (one where the atoms are close together), particularly if the atoms in a single rigid body straddle a periodic boundary. This means the input data file or restart file must define the image flags for each atom consistently or that you have used the set command to specify them correctly. If a dimension is non-periodic then the image flag of each atom must be 0 in that dimension, else an error is generated.