Hello,
I’m simulating a rigid mixture of CO2 and Acetonitrile (primarily ACN, with a little bit of CO2). I’ve started by running phase equilibria calculations in another program (GOMC, using Gibbs Ensemble Monte Carlo), and then I have been starting from the compositions calculated from those calculations and running lammps MD simulations to calculate the diffusion coefficient. Eventually, I want to include ions in the calculation, but I’m currently working on the simple binary system. The densities that I am calculating from lammps match well with my GEMC calculated densities.
I’m getting a strange behavior in the diffusion coefficient, where for some portions of the trajectory the calculated mean-squared displacements have a linear regime (slope of 1 on a log-log plot) and for other portions it has a non-linear regime (slope of like 1.3 on a log log plot). There doesn’t seem to be a clear indicator to me of what the cause might be. I’ve experienced this issue on both the 7Aug2019 version of lammps, as well as the 3Mar2020 version. This behavior, when it happens, typically occurs later on in the trajectory after quite a long period of time (usually sometime over a nanosecond into the simulation). I’ve looked through the mailing list and it seems the general advice for fix rigid in these scenarios is use a smaller timestep; however, as I mention below, that didn’t fix my problem. Both of my molecules are linear, 3 site models, so fix SHAKE isn’t an option in this case.
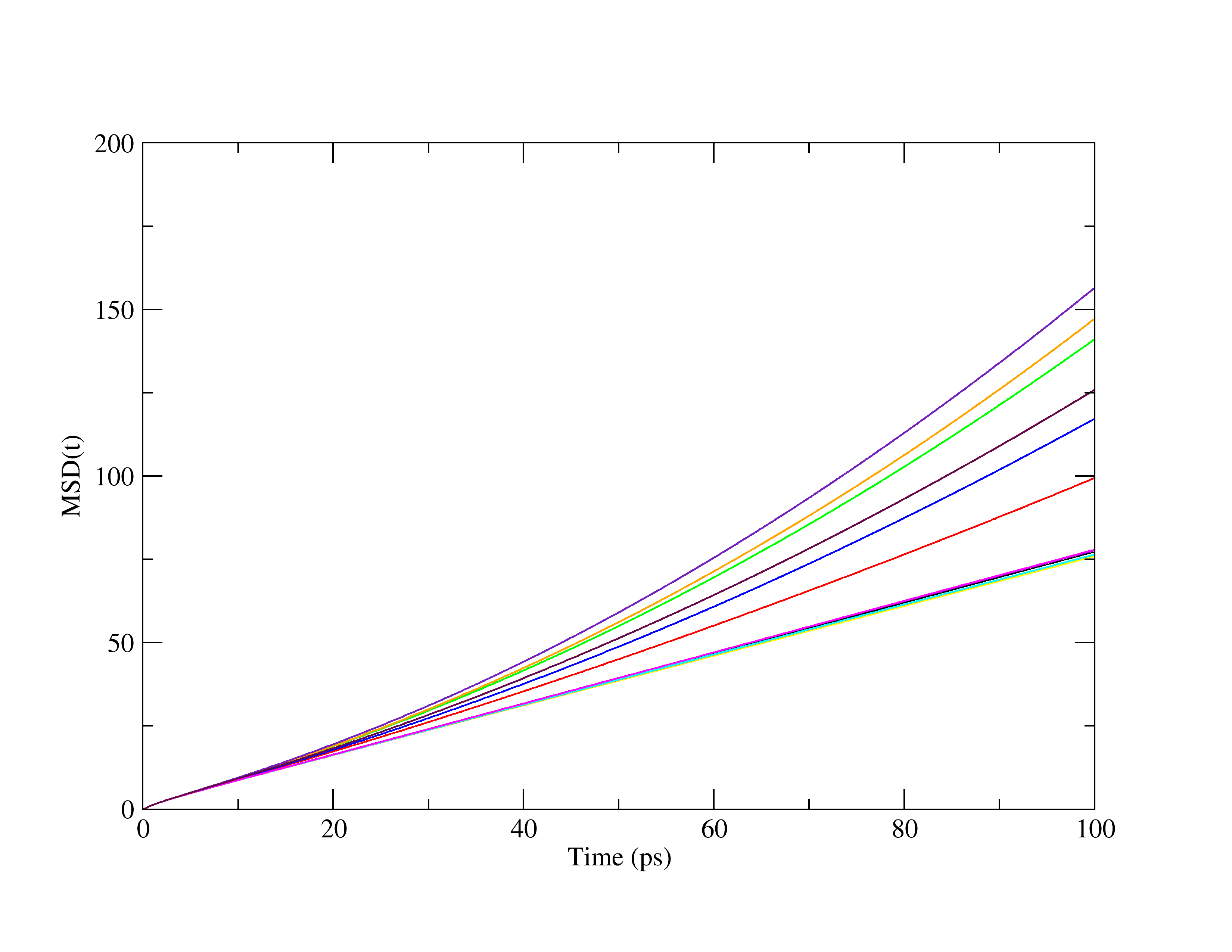
I’ve included a plot of the MSD versus time calculated for 2 ns chunks from my simulations, as you can see, the grouping on the bottom is linear as expected, the grouping on the top is very much not.
Things I’ve tried:
- Doubling/halving the system size
- Halving the simulation timestep from 1.0 fs to 0.5 fs
- Tried running with both rigid/nvt and rigid/nvt/small variants
- Running with a separate rigid fix for each molecule type, instead of a single rigid fix for both
- Doubling the output frequency of the dump/outputting in wrapped/unwrapped coordinates (to eliminate coding issues with the msd calculation)
- My MSD code works for non-rigid systems in reproducing the diffusion coefficient
I’ve attached a sample set of input files a binary simulation test set that I have been using, with 999 ACN molecules and 1 CO2 molecule. The test set that I am providing has no ions in it, but does my whole procedure of 500 ps of NVT equilibration, 1 ns of npt equilibration after which it calculates the average box size for 1 ns, and then a 500 ps nvt equilibration again at the new box size before a 5 ns production run (I include it all in case there is an issue with using change-box with fix rigid or some other problem I might be overlooking). I would appreciate if someone had a suggestion for more tests that I could pursue to trying and figure out the cause of the problem, or if this is something anyone has seen before?
I’ve been challenged by this for a few weeks, so I really would appreciate any insight into this that anyone might have! Thanks so much in advance! I can also supply my MSD code if that would be helpful, but I don’t expect anyone to want to look through it (and at this point I’m leaning towards the problem being with something I’m doing in my simulations rather than in my code).
Best,
Zeke
data.lmps.gz (78.2 KB)
lmps.include.coeffs.gz (342 Bytes)
in.acncx.gz (758 Bytes)