Dear all,
I am confused when I’m using the command fix wall/region with a union region. For example, two block regions are defined as:
region 1 block 0 100 0 100 0 10 units box
region 2 block 45 55 45 55 11 50 units box
region substrate union 2 1 2 units box side out
the region substrate is used by the command fix wall/region as:
fix wall all wall/region substrate lj93 0.03 2.5 20.0
particles are initially created in the region substrate, i.e., no particles are in block region 1 or 2.
I have two questions:
-
there are convex edges/points felt by particles, but they are not additional sharp edges due to the intersection, can lammps handle this kind of edges correctly?
I notice the sentence in the document "
as a general rule you should not use the fix wall/region command with union or interesect regions that have convex points or edges.", so I want to make sure I understand this correctly.
-
what’s the difference if I define region 2 as: region 2 block 45 55 45 55 10 50 units box.
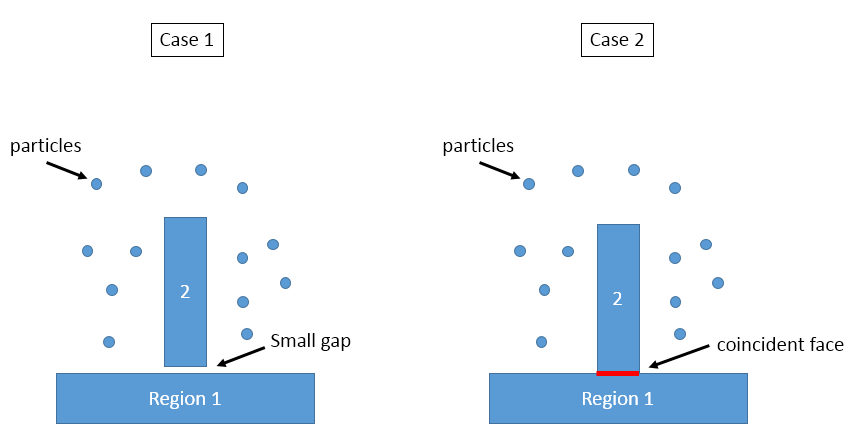
you may notice that previous region 1 and 2 have a small distance 1 in z direction, this is following the suggestion: “Having two s could cause the face to become invisible to the particles. The solution is to make the two faces differ by epsilon in their position.” but I think the distance between region 1 and 2 in z direction is just 1 Angstrom, no particles will penetrate into this small gap, so the forces act on the particles by the wall substrate is quite the same. Anyone have some comments on this?
Best,
Wei Xiong
Have you tried running and visualizing a simulation to see
what happens?
Steve
I’ve run 3 testing simulations, no differences were found (thermodynamics output are the same) for the following 3 settings.
I can understand why case 1 and case 2 are the same, but I do not understand why case 2 and case 3 are also the same. For a particle have a distance r from both surfaces of block 0 and block 1, the potential energy is lj93(r) in case 2, but 2*lj93(r) in case 3, why the thermodynamics output are the same?
- two block regions have a small gap in z direction:
region 0 block 1.0 199.0 1.0 199.0 -10.0 0.0 units box
region 1 block 75.0 125.0 75.0 125.0 1.0 60.0 units box
region substrate union 2 0 1 units box side out
fix wall all wall/region substrate lj93 0.03 2.5 20.0
fix_modify wall energy yes
- two block regions have a coincident face:
region 0 block 1.0 199.0 1.0 199.0 -10.0 0.0 units box
region 1 block 75.0 125.0 75.0 125.0 0.0 60.0 units box
region substrate union 2 0 1 units box side out
fix wall all wall/region substrate lj93 0.03 2.5 20.0
fix_modify wall energy yes
- multiple fix wall/region are used:
region 0 block 1.0 199.0 1.0 199.0 -10.0 0.0 units box side out
region 1 block 75.0 125.0 75.0 125.0 0.0 60.0 units box side out
fix wall0 all wall/region 0 lj93 0.03 2.5 20.0
fix_modify wall0 energy yes
fix wall1 all wall/region 1 lj93 0.03 2.5 20.0
fix_modify wall1 energy yes

I don’t know. I think the only way to debug union
regions with this kind of complexity is to use a single
particle placed at an interesting position, then
print out the force on the particle to see if it
is what you expect. You may then need
to put some print lines in fix_wall_region.cpp to see
what “closest to surface” points are returned.
Steve
I want to know, in case 3 with multiple fix wall/region, the potential energy for a particle with a distance r0 from region 0 surface and r1 from region 1 surface, is lj93(r0)+lj93(r1)? Is this the way LAMMPS works?
在 2014年8月22日,22:51,Steve Plimpton <[email protected]> 写道:
I want to know, in case 3 with multiple fix wall/region, the potential
energy for a particle with a distance r0 from region 0 surface and r1 from
region 1 surface, is lj93(r0)+lj93(r1)? Is this the way LAMMPS works?
this is the wrong question. set up a test with a single test atom and
see for yourself: put the atom in all kinds of places and compute
energy and forces manually and with LAMMPS and compare. that will give
you a definite answer.
axel.
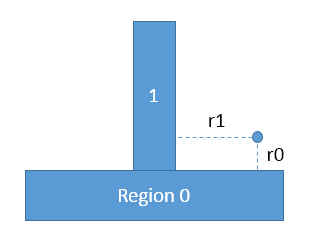
Steve and Axel, I followed your advice, put a single atom in some fixed positions and compare the forces and energy calculated manually and by lammps. The results in the following two cases are exactly the same, which is wired to me. Lammps outputs are consistent with the results calculated manually for case 3.
For example, please see the attached sketch, I can not understand why there will be a force fx in case 2. I thought the potential energy of the atom is lj93(r0) in case 2, lj93(r0)+lj93(r1) in case 3. Because in case 2 with a union region, the atom interact with the wall as a whole, so it finds the nearest point with a distance r0 (suppose r0<r1). But in case 3 with multiple fix wall/region, the atom interact with two walls, so it finds the nearest point separately.
But Lammps seems not to work this way. For settings in case 2, lammps gives output exactly the same with that in case 3. Can this be a bug? Or can you please explain more details how lammps deal with union region like this?
Wei Xiong
case 2: union region
region 0 block 1.0 199.0 1.0 199.0 -10.0 0.0 units box
region 1 block 75.0 125.0 75.0 125.0 0.0 60.0 units box
region substrate union 2 0 1 units box side out
fix wall all wall/region substrate lj93 0.03 2.5 20.0
fix_modify wall energy yes
case 3: multiple fix wall/region are used:
region 0 block 1.0 199.0 1.0 199.0 -10.0 0.0 units box side out
region 1 block 75.0 125.0 75.0 125.0 0.0 60.0 units box side out
fix wall0 all wall/region 0 lj93 0.03 2.5 20.0
fix_modify wall0 energy yes
fix wall1 all wall/region 1 lj93 0.03 2.5 20.0
fix_modify wall1 energy yes
 --------------------> x direction
--------------------> x direction
Steve and Axel, I followed your advice, put a single atom in some fixed
positions and compare the forces and energy calculated manually and by
lammps. The results in the following two cases are exactly the same, which
is wired to me. Lammps outputs are consistent with the results calculated
manually for case 3.
For example, please see the attached sketch, I can not understand why
there will be a force fx in case 2. I thought the potential energy of the
atom is lj93(r0) in case 2, lj93(r0)+lj93(r1) in case 3. Because in case 2
with a union region, the atom interact with the wall as a whole, so it
finds the nearest point with a distance r0 (suppose r0<r1). But in case 3
with multiple fix wall/region, the atom interact with two walls, so it
finds the nearest point separately.
no, that is not how i read the documentation. LAMMPS will consider each
face separately and compute the force accordingly. your observation is thus
consistent with the description and correct. i would consider your case 2
behavior as inconsistent and unexpected instead.
axel.

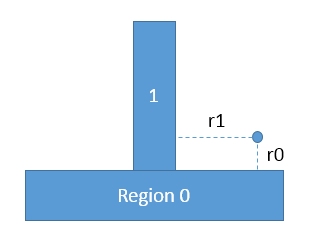
Axel, thank you for your clarification. If lammps consider each face (in
primitive regions and union regions) separately, I can understand why
lammps gives the same results for my testing case 2 and case 3.
But what do you mean by saying "
i would consider your case 2 behavior as inconsistent and unexpected instead"?
In case 2, as you said, lammps consider each face in the union region
separately, so the potential energy for the particle should be
lj93(r0)+lj93(r1) (cf. the attached sketch), the same with that in case 3,
lammps gives consistent results with this explanation.
Wei Xiong
*case 2: union region*
*region 0 block 1.0 199.0 1.0 199.0 -10.0 0.0 units box*
*region 1 block 75.0 125.0 75.0 125.0 0.0 60.0 units box*
*region substrate union 2 0 1 units box side out*
*fix wall all wall/region substrate lj93 0.03 2.5 20.0*
*fix_modify wall energy yes*
*case 3: multiple fix wall/region are used:*
*region 0 block 1.0 199.0 1.0 199.0 -10.0 0.0 units box side out*
*region 1 block 75.0 125.0 75.0 125.0 0.0 60.0 units box side
out*
*fix wall0 all wall/region 0 lj93 0.03 2.5 20.0*
*fix_modify wall0 energy yes*
*fix wall1 all wall/region 1 lj93 0.03 2.5 20.0*
*fix_modify wall1 energy yes*
!333.png|311x238--------------------> x direction
Axel, I may need your further comments about this topic to make it more clear. Is the potential energy of the single particle in my testing case 2 and 3 are both lj93(r0)+lj93(r1)? Thank you for your time.
在 2014年8月24日,21:35,Wei Xiong (熊伟) <xwwell@…24…> 写道:
Axel, I may need your further comments about this topic to make it more
clear. Is the potential energy of the single particle in my testing case 2
and 3 are both lj93(r0)+lj93(r1)? Thank you for your time.
if you want a definite answer, you have to read through the source
code (as would i).
Axel, thank you for your suggestion.
I’ve read the code fix_wall_region.cpp, region.cpp and region_block.cpp, lammps does consider each face in the region separately, so it is consistent that lammps gives the same results for my testing case 2 and 3. In other words, I believe the potential energy for the single particle should be lj93(r0)+lj93(r1) for case 2 and 3.
May be it is a good idea to add some explanation like “lammps consider each face in the region separately” in the documents.
Yes, I think your diagram of the exterior of the union of 2 blocks is effectively no
different than what LAMMPS does for the interior of a single block
(for the case of a particle near the corner of the 2 blocks, as in your
diagram).
I.e. for a single block, if a particle is near a corner (interior), the particle
can feel forces from 1,2,or 3 planar walls, depending on the distance from
each. So LAMMPS calculates all those distances and applies 1,2, or 3
force components. A sphere region only has one possible force.
A cylinder region has 2 where a particle is near the end cap, might feel force
from the side wall and the end cap. I.e. each primitive region creates one
or more possible force components. For a union/intersect region the lists
from each primitive region are combined, and I think list entries are discarded
if they originate from a point that is discarded by the union or intersection operation.
Steve
May be it is a good idea to add some explanation like “lammps consider each face in the region separately” in thedocuments.
yes, I will add some text about this,
thanks,
Steve
{kind=link}