Dear all,
LAMMPS Model being used: 2024-03-02 18:23 LAMMPS-Win10-64bit-GUI-stable.exe
I am attempting to follow a water box guide from Polymer in water -
I have managed to run the file after altering the code slightly.
The code I changed was the following:
molecule h2omol H2O-SPCFw.mol
create_atoms 0 random 1050 87910 NULL mol &
h2omol 454756 overlap 1.0 maxtry 50
Changed to:
molecule h2omol H2O-SPCFw.mol
create_atoms 0 random 1050 87910 NULL mol h2omol 454756 overlap 1.0 maxtry 50
When I try to view the dump file the hydrogen and oxygen molecules are not bonded in a proper manner. Below is a picture of what it looks like.
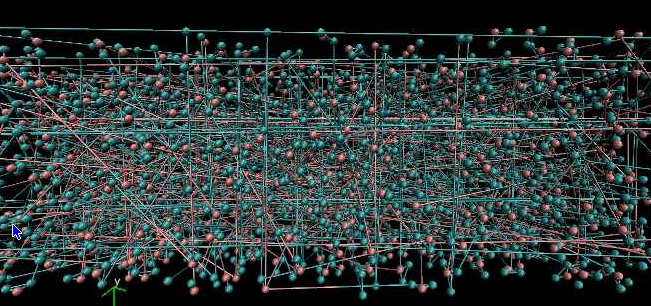
I will also provide all the code I am using to run the simulation here.
units real #masses are in grams per mole, distances in Ångstroms, time in femtoseconds, energies in Kcal/mole.
atom_style full #each atom is a dot with a mass and a charge that can be linked by bonds, angles, dihedrals and/or impropers
#bond_style, angle_style, and dihedral_style commands define the potentials for the bonds, angles, and dihedrals used in the simulation, here harmonic.
bond_style harmonic
angle_style harmonic
dihedral_style harmonic
pair_style lj/cut/coul/long 12 #atoms interact through both a Lennard-Jones (LJ) potential and Coulombic interactions. The value of 12Å is the cutoff.
kspace_style pppm 1e-5 #defines the long-range solver for the (long) Coulombic interactions. The pppm style refers to particle-particle particle-mesh
special_bonds lj 0.0 0.0 0.5 coul 0.0 0.0 1.0 angle yes #cancels the Lennard-Jones interactions between the closest atoms of the same molecule.
#create a 3D simulation box of dimensions 9x3x3nm^3, and make space for 9 atom types (2 for the water molecule, and 7 for the polymer molecule)
#7 bond types, 8 angle types, and 4 dihedral types.
region box block -45 45 -15 15 -15 15
create_box 9 box &
bond/types 7 &
angle/types 8 &
dihedral/types 4 &
extra/bond/per/atom 3 &
extra/angle/per/atom 6 &
extra/dihedral/per/atom 10 &
extra/special/per/atom 14
#file contains all the parameters (masses, interaction energies, bond equilibrium distances, etc). types 8 and 9 are for water and the atoms of types 1 to 7 are for the polymer molecule
include PARM.lammps
#create water molecules
molecule h2omol H2O-SPCFw.mol #molecule template
create_atoms 0 random 1050 87910 NULL mol h2omol 454756 overlap 1.0 maxtry 50
group H2O type 8 9 #organize the atoms of types 8 and 9 of the water molecules in a group named H2O
minimize 1.0e-4 1.0e-6 100 1000
reset_timestep 0
fix mynpt all npt temp 300 300 100 iso 1 1 1000
#print the atom positions in a .lammpstrj file every 1000 steps (i.e. 1 ps), print the temperature volume,
#and density every 100 steps in 3 separate data files, and print the information in the terminal every 1000 steps
dump mydmp all atom 1000 dump.lammpstrj
variable mytemp equal temp
variable myvol equal vol
fix myat1 all ave/time 10 10 100 v_mytemp file temperature.dat #print the temperature
fix myat2 all ave/time 10 10 100 v_myvol file volume.dat #print the volume
variable myoxy equal count(H2O)/3
write_data H2O.data