It seems there are some inconsistency in the formation energy of sulfides and fluorides.
For example, the formation energy of Li2S is -4.681 eV (-1.56 eV/atom) in reaction calculator, but is -1.339 eV/atom in mp-1153. This problem is noticed for fluoride also. Similar search on oxides and others would give consistent results.
It seems that the recent S anion correction is not included for the formation energy in the sulfide at the MP entry pages.
It turns out that when we performed updates for the sulfide correction, we updated energy-above-hull values but not formation energies. Now, formation energies should all be updated as well. Thank you for noting the inconsistency.
I also have questions about formation energies. I don’t know how the formation energy is calculated in Materials Project. Why is it listed as eV/atom? Is it just simply the formation energy per chemical formula divide by the number of atoms? In the Li2S case, it is just -4.681/3=-1.5603333 eV/atom? If this is the case, then I should say the formation energy of RuO2, for all the structures that on Materials Project seem not correct. I am looking forward to your reply.
Our formation_energy_per_atom [1] value is the energy of the compound with respect to standard states (elements), normalized per atom. e.g., for Fe2O3 this is: [E(Fe2O3) - 2E(Fe) - 3/2E(O2)]/5.
It is computed at 0K, 0atm using a reference state of zero for the pure elements. This quantity is often a good approximation for formation enthalpy at ambient conditions. [2]
Thank you for looking into my previous problem. It is fixed.
Now I note some more inconsistencies of the E_above_hull and the phase diagram construction.
E.g. If you look at
S2N mp-2565: the entry pages gives a E_above_hull of 0.303 eV. But the same phase is shown as stable in the Phase Diagram App as quite favorable.
Mn2S3 mp-974355: the entry pages give a E_above_hull 0f 0.000 eV. But the Phase Diagram App shows the same phase/composition as metastable.
It seems the bug is largely associated with systems that two elements (O, S, N, halogens, H, TMs) need to be corrected. For example, many mixed anion compounds seem to have this issue, so as some H+anion and TM+anion systems.
It seems the issue being that multiple correction terms (when two or more are needed) are not applied consistently somehow?
I am trying to reproduce the results for pure Silicon in different space groups.
I assume that in the pure element case, what is listed in MP is the Formation energy (FE) wrt the most stable structure available, since the first value is always 0.
The lowest Si structure reported is spacegroup Fd-3m (227, mp-149) with FE=0, the second one is mp-165 with FE=0.011 and the third is mp-971662 with FE=0.063 (all values in ev/at)
According to my calculations (from pw.x, volume and density agree) FE(mp-149) = -5.6500, FE(mp-165) = -5.644 and FE(mp-971661)=-5.5982 eV/at.
The difference in the two first is, FE(mp-165)-FE(mp-149)=0.0057, which is one half of the MP result, and I don’t know how to justify this since all results are per atom. FE for the second case (FE(mp-971661)-FE(mp-149)=0.051785, while MP result is 0.063.
Also, there are two occurrences of the same spacegroup (Fd-3m, or 227), one is mp-149, the other is mp-16220. The first one has FE=0, the second FE=0.34 ev/at. The only difference being the number of sites (2 vs.34). How is this value estimated?
thank you
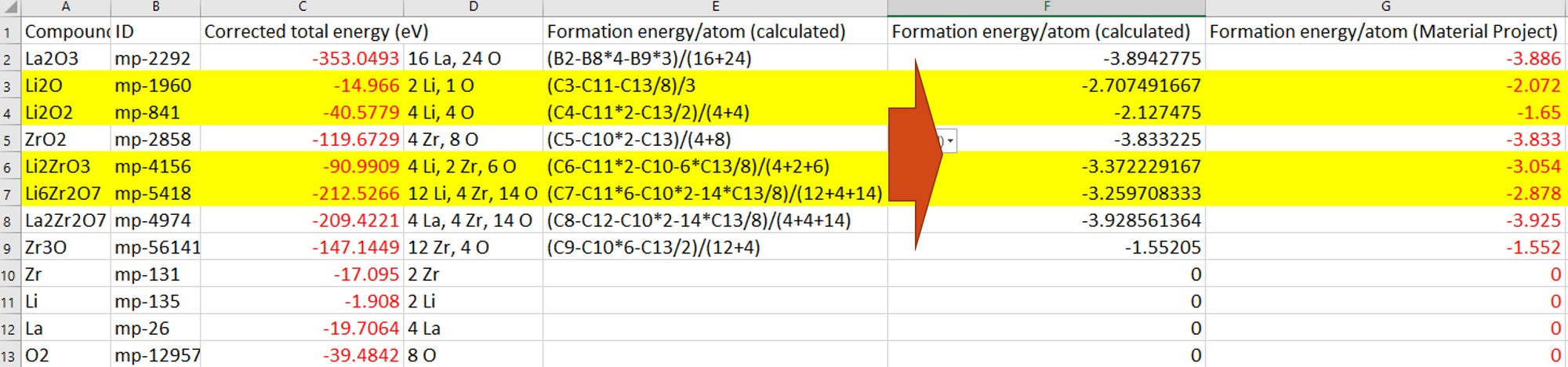
I have question related to the formation energy calculation. I find some inconsistency between formula calculation and Material Project value for several compound such as Li2O, Li2O2, as shown in the figure. Could you please help to verify which values is exactly?
The following is the updated table.It seems to me that the compound related to Li has problem with inconsistency formation energy. Or did I made some wrong calculation? The “red” values are taken from Material Project.