I’m having some trouble with my simulation. My goal is to keep one group of atoms still (I’m calling this the freeze group) so they remain the same (similarly) without breaking or distortion during high-pressure while letting another group freely move (the mobile group). I’m using both NVT and NPT ensembles, adjusting the NPT in the z-direction only. However, I’m struggling to apply NPT correctly to both the freeze and mobile groups in my triclinic system.
First Try - Using fix rigid
fix 1 mof rigid/nvt single temp 300 300 100
fix 2 mobile npt temp 300 300 100 z 592 592 1000 dilate all
: The problem is, that the simulation box isn’t adjusting as expected. For example, I expected it to shrink to around 100Å, but it shrunk to 300Å instead.
Second Try - Trying to Immobilize
fix rigidMof mof rigid single
fix freeze mof setforce 0 0 0
velocity mof set 0 0 0
: The simulation box size adjusted reasonably, but then I got an “ERROR: Dihedral atoms missing”.
I’ve attached the detailed input scripts for my system. Could you please look them over and tell me what I might be doing wrong? Any suggestions on how to fix these issues would be greatly appreciated.
You cannot have two ensembles at the same time, in fact, your simulation is in neither of the two.
See the “guidelines and suggestions” post for a discussion of this topic.
This has been discussed many times and there is no “clean” or “correct” solution for it. When you use an NPT fix you can choose to either rescale all positions according to the adjusted box or a subset. As a consequence your immobilized atoms are either moving (because they were rescaled, too) or they stay in place and then their distance to the rescaled positions of the other atoms changes; in extreme cases the mobile atoms are moved so far by the rescaling that they will result in unphysical overlaps or gaps in the system.
Different people have voiced different opinions about what is the best approach (or rather the least bad). You can find these discussions in the archives.
I know what you are trying to do. I also failed to do this 21-step-equilibration protocol 48390248 times at the time I was trying a couple of years ago.
About try1.in:
Your box is not shrinking as expected because the pressure calculation is bogus when you dont turn off the interactions inside the rigid body, and this is going to be a problem in the NPT stage since the dynamics and the changing of the box dimensions rely on teh calculation of pressure. You can turn these off for the mof using the commands:
neigh_modify exclude group mof mof check no
delete_bonds mof multi
If you check in the manual you will be able to figure out what it is doing. Note that since you are turning all the interaction between mof atoms off, you will need to do something about it before you start doing some “proper” dynamics after this 21-step-cycle (I, myself, simply start a new simulation once I finish it).
Other than that, I liked in principle the way you arranged each set of 3 steps in the script. I think it is reasonable within what the protocol prescribes. But in fact there is an additional problem, and this one I do not know very well how to explain to you. I would need to read some of my old notes and check again carefully the manual page for these commands. I noticed it was also necessary to have, at least during the NPT stage, a fix 2 mof setforce 0.0 0.0 0.0 command, so that it would look something as shown below. In my case I simply kept the MOF as a rigid block, moving naturally as a whole simply as a consequence of the shrinking of the simulation box, while the polymer is merely “equilibrating under its presence” - in principle your approach of moving via a nvt/rigid seems reasonable, but since you are going to set the force to 0, it wouldnt make sense to try do some “single-block” dynamics.
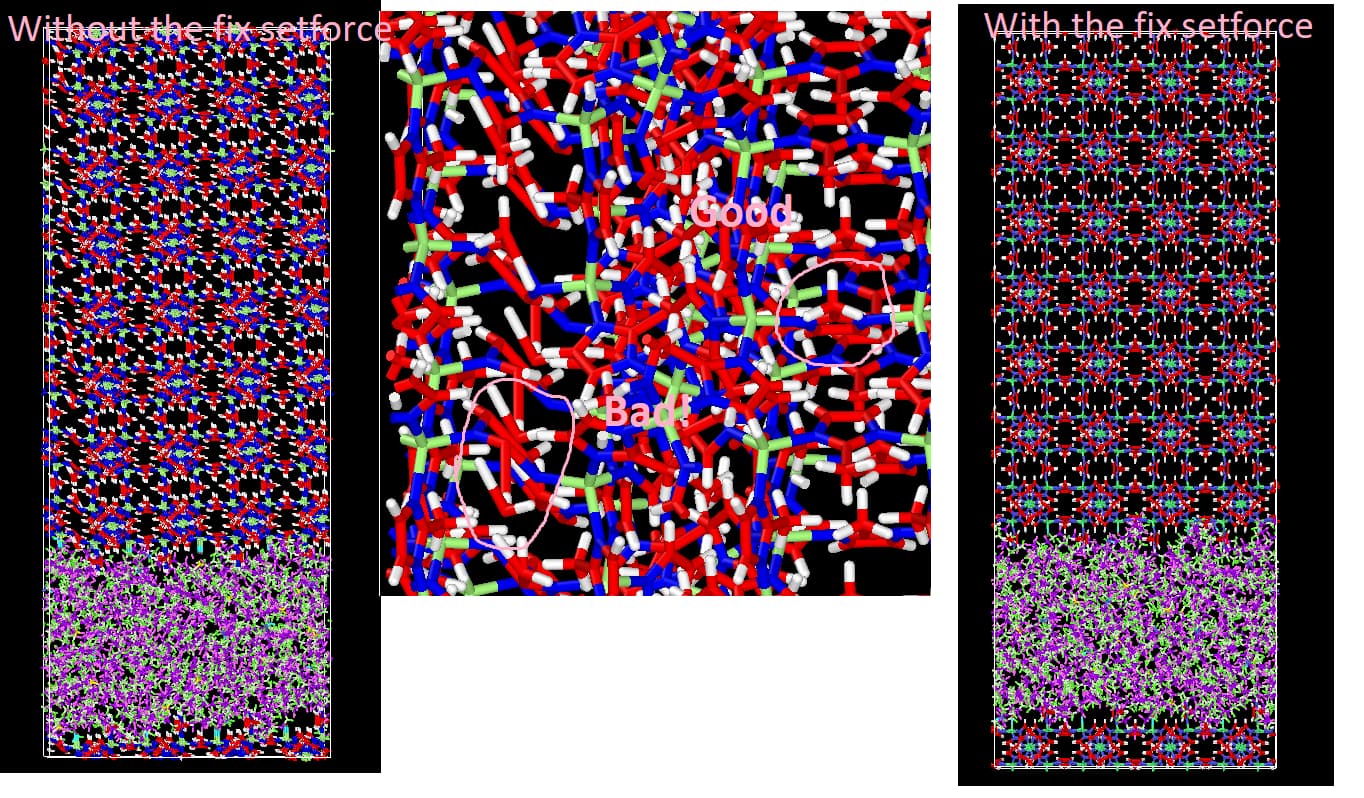
I dont know why you need to set this fix force command - this is the thing I would need to check my old notes and the manual again in order to try to reason an explanation. In principle, since you are not really applying any equations of motion to it and the body is going to move as a whole no matter what happens to the simulation box, it seems to me now that it should simply keep the MOF as it originally was in terms of relative positioning of the atoms. But I can assure you that if you dont apply the fix setforce, something weird happens for some reason. For example, I can show you below how my MOF phase looked like in the data file saved after the 21-steps, with the NPT designed as shown above and with the NVT slightly different than yours (but I think your approach to it work just fine), if it is carried out without using the fix setforce - and particularly, if I track back, the problem started in the first NPT stage. My MOF is ZIF-8, where the linkers are methyl-imidazole rings, and you can see that some of them are not exctly looking good (i.e., planar). If you use the fix setforce, no weird things happen (you can see also that the block is not tilted, as it is in the first case, which I think should be the most reasonable scenario in this case since I see no reason for it to tilt in this context).
About try2: I think this one would not make much sense within LAMMPS, because you are defining the MOF to be rigid and also using a fix nvt all, which naturally would encapsulate the MOF atoms, and these are contrasting command lines since the latter would want to apply the usual Nose Hoover equations of motion (originally, but not here) used to sample the NVT ensemble to the individual atoms. So try2 you can forget, it will not work.
By the way, you should not need this “check no” that I put in my neighbor_modify command unless you enjoy taking 3 additional years to run your simulations like I apparently did. (I think this should significantly slow down everything compared to the check yes, so removing it is not a bad idea)
Thank you for your quick and kind response.
What I meant by “I’m using both NVT and NPT ensembles” is that I equilibrate the system through three successive steps: 1) NVT at 600K, 2) NVT at 300K, 3) NPT.
I am trying to find the best approach for my system.
Thanks a lot for your advice!
Thank you for your kind response!
It seems that your system is very similar to what I am working on. Your suggested approach works well with my system too. I really appreciate your help!
Just one last thing: I would advice you to do not get out of this 21-step cycle and consider that your system is “equilibrated” in the sense that you are going to straightforwardly derive results from the dynamics that happens right after. Your system may not yet be equilibrated and thus you need to have a “physical” equilibration stage just for any other simulation. The idea of the 21-step-cycle is not exactly to “equilibrate” the system in the orignal sense. It is merely an adaptation of a procedure previously developed for polymer systems alone to converge to an initial configuration believed to be “physical” for starting some meanigful simulations in a scenario where you had a configuration built artificially. This procedure, as well as this one for the MOF/polymer system, is particularly useful for when the polymer is very rigid, which would cause a very high simulation time in “usual” (T,p) conditions to be needed (rigid polymers in general may already be difficult to study per se in the sense that you may not be able to explore in a representative way the full conformational space with a “reasonable” amount of timesteps).
So bottom line is that the procedure does not really replace an equilibration, just “is believed” to lead to a reasonable initial configuration to start doing the whole equilibration + production simulation.