Hi. I’m trying to understand the physical role of the global interactions cutoff in the GB potential. I know that, when specifying the global interactions cutoff, I have to chose a distance that is greater than the largest ellipsoidal axis of the system. I am simulating a system of active ellipsoids and spheres, setting the parameters for ellipsoids as 3.0 1.0 1.0 and 1.0 1.0 1.0 for spheres, and therefore presume the cutoff can be specified in the range of >3?
Currently, I’m specifying 2 pair coefficients: one for ellipsoids-ellipsoids and one for sphere-sphere interactions as follows. I use mix geometric for ellipsoids-sphere interactions:
However, LAMMPS documentation mentions the local cutoff variable, and I get different potential energy results when I specify local cutoffs for spheres and ellipsoids differently, versus just specifying the global cutoff, and my simulations behave differently in general. Can someone give an insight into this, on the role of the cutoff parameter, or is there something wrong with my coefficients set up? Thank you, really appreciate your help!
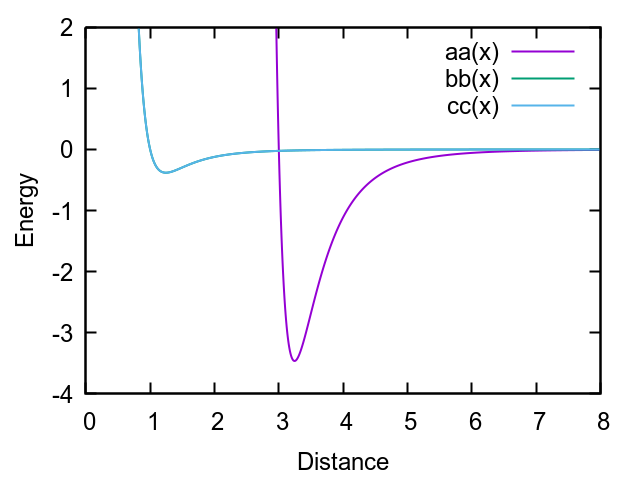
The way you defined the GB potential is not very physical. For a prolate object, the deepest energy well is that for the y and z direction, in your convention. But with the parameters you have specified, this is what you get:
This potential describes an elongated object (a prolate) that is very attractive at the tips and very shallow on the side. Very wired, if you ask me.
A cut-off of 3 will result in a bad conservation of energy, which is clear from the shape of the potential.
The convention mutated from the liquid crystal community is to assign the optical axis to z: this means that for an oblate (a disk) z is the shortest axis, while for an oblate z is the longest. Also, the pair_coeff uses an eps0 to set the energy well. I have always assumed that this implies setting the parameters epsx, epxy, and epsz in the range (0:1], as in mixed interactions the parameter eps0 is combined, while the relative well depths remain the same for each particle type.
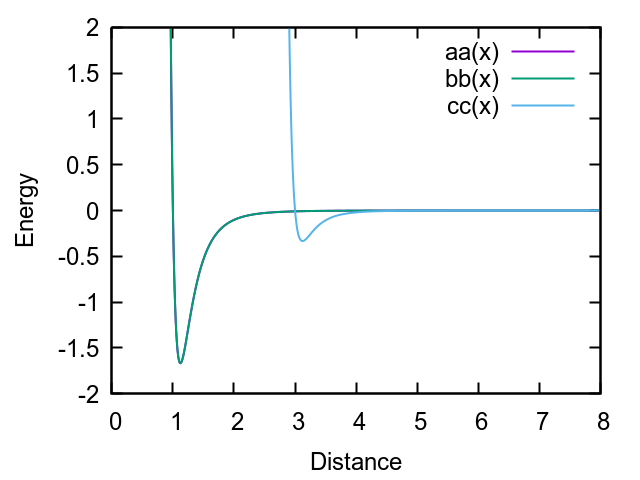
You may want to experiment with the following parameters:
set type 1 shape 1.0 1.0 3.0
pair_style gayberne 1.0 1.0 1.0 4.5
pair_coeff 1 1 3.0 2.0 1 1 0.2 1 1 0.2 # Edit: I made a typo in the relative well depths.
pair_modify mix geometric
Which gives the potential:
Please also search the forum for similar discussions on the Gay-Berne potential. It always helps to visualise the potential, so I leave here the Gnuplot script that I have used to make the plots. gayberne.gnu (934 Bytes)
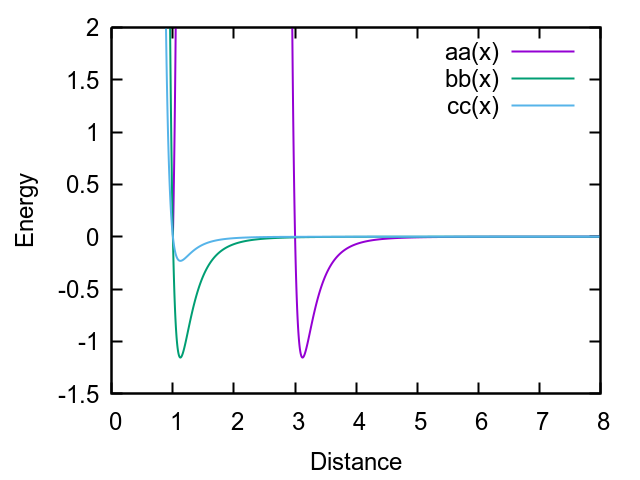
The script examples/ellipse/in.ellipse.gayberne, crafted by Steve Plimpton himself[1], is a bit misleading as it uses the following parameters:
set type 2 shape 3 1 1
pair_coeff 2 2 1.0 1.0 1 1 0.2 0 0 0
resulting in the following Gay-Berne potential:
For a uniaxial ellipsoid (3 1 1), one would expect the bb and cc curves to be the same. While this is not an error per se, I would suggest following the uniaxial convention:
Thank you for your guidance here, especially for sharing the Gnuplot script - that is very insightful! I think I have a better understanding about the 3D case, but I am still struggling to understand how to set up the Gay Berne potential if I am using 2D simulations? Since I wouldn’t be able to “elongate” the ellipsoids in the z-direction. Would it look something like:
set group active type 1 shape 1.0 3.0 1.0
set group passive type 2 shape 1.0 1.0 1.0
Also, you mentioned that ellipsoid parameters (epsx, epsxy, epsz) should be within the range 0:1 - would it mean that 3.0 parameters would be replaced by 1.0? Thank you in advance for your reply and patience!
In 2D simulations, the equation of motions are only integrated in 2 dimensions, but the objects remain fully 3D. See the examples in lammps-YOURVERSION/examples/ellipse/.
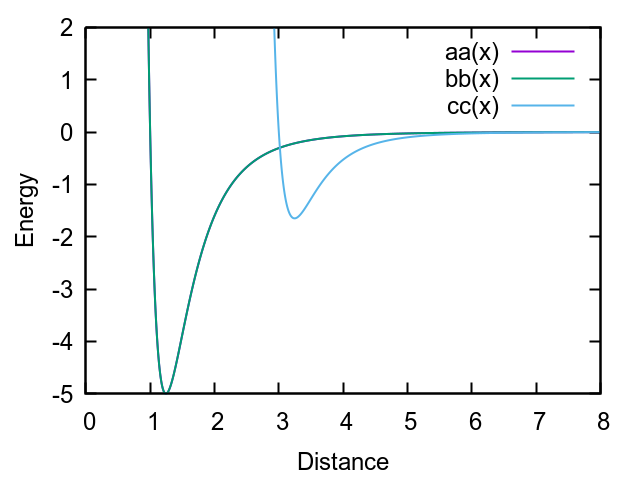
The energy well depth is controlled by the parameter eps0. Please compare the pair coefficients:
Besides the difference in sigma0 (which has been discussed here), in my definition the well depth is eps0 = 3.0.
I have noticed that the gnuplot script that I posted in my previous reply used absolute well depths[1] instead of relative ones, as LAMMPS does. This is the amended potential curve:
And the updated script: gayberne.2.gnu (959 Bytes)
Which I use for fitting the Gay-Berne potential. ↩︎