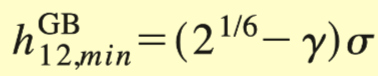Perhaps somebody who is more familiar with the Gay-Berne potential in LAMMPS can reply to your question more directly.
The paper describing the generalized Gay-Berne potential used in LAMMPS is here:
http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.78.3137&rep=rep1&type=pdf
In case nobody else replies, and if you cannot figure it out, then perhaps you can measure the position of the minima yourself by measuring the energy as a function of distance between the two particles. Here are some suggestions how to do that using LAMMPS:
-
First, please download the latest version of LAMMPS from github.
There was a bug fix to pair_style gayberne.
It’s possible that versions of LAMMPS which are older than a few days may be calculating the Gay-Berne interaction incorrectly. (See here for details.)
-
Try placing two atoms at different distances from each other and measure the potential energy. Probably the easiest way to do this is to create many LAMMPS data files. Each data file contains only two particles. In each file place the two particles at a different distance from each other.\ Then use the “read_data” command to read these data files and run a short simulation. Run a short (1-timestep) simulation and print out the potential energy of the system at the end of the simulation (using the “thermo” or “print” commands).
-
Documentation:
There is a simple explanation of the LAMMPS data format here:
https://lammps.sandia.gov/doc/2001/data_format.html
However this old documentation does not explain how to include ellipsoids in your data file.
However there is an example of a LAMMPS data file containing ellipsoids here:
https://github.com/lammps/lammps/blob/master/examples/USER/misc/ees/Data_region
Try modifying this file (get rid of the “Velocities” section, get rid of the 3rd line in the “Atoms” and “Ellipsoids” section. Then edit the last 3 columns of the “Atoms” section to control the positions of the two atoms. You can edit the last 7 columns of the “Ellipsoids” section to control the shape and orientation of the ellipsoids.
There’s an example of a LAMMPS input script that uses ellipsoids here:
https://github.com/lammps/lammps/blob/master/examples/ellipse/in.ellipse.gayberne
My apologies if this is not the reply you were expecting.
Hope this helps.
Andrew
P.S. Also take a look at the “pair_write” command.
https://lammps.sandia.gov/doc/pair_write.html
I hesitate to suggest this command because I doubt that it will work when using ellipsoidal particles. (That’s why I suggested making a series of LAMMPS data files instead and running short simulations instead.) More random documentation:
https://lammps.sandia.gov/doc/read_data.html
https://lammps.sandia.gov/doc/run.html
https://lammps.sandia.gov/doc/print.html
https://lammps.sandia.gov/doc/thermo.html
P.P.S. There’s another example of building a data file containing ellipsoids here:
http://moltemplate.org/visual_examples.html#ellipsoids
…but that example requires that you have moltemplate installed.