GCMC with Electrode (conp)
Hi Everyone,
I have been trying to simulate GCMC adsorption of ions in a nanotube which act as metal-like walls. For the same I was trying to use both GCMC and the Electrode package in combination but with no success. Below I explain my procedure in detail. You can find my simulation files and lammps edits here.
1. Lammps Modifications
The primary focus of the lammps modifications was to repeoduce the energy calculations conducted by GCMC (FixGCMC::energy_full()) by obtaining the system state from inside the FixGCMC::energy_full() and running it as a new identical case but using nvt (only to compare the energy without changing the coordinates). For the same, the following changes are made to the lammps (2Aug2023_update2) source files (see the lammps folder here).
1.1 GCMC State
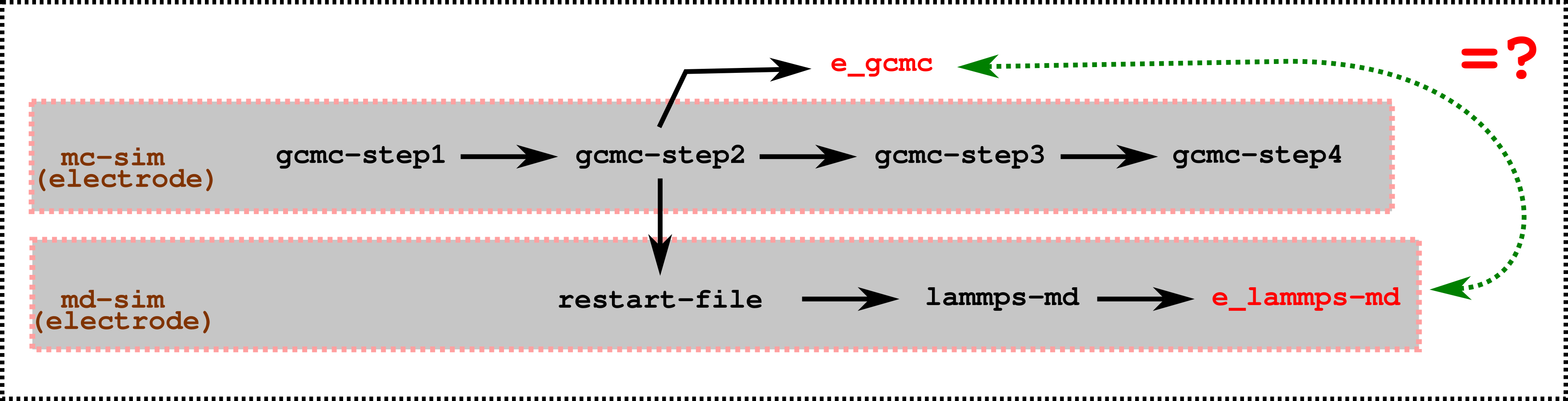
For getting the state of the system during the FixGCMC::energy_full() call, I add a output->write_dump() and output->write_restart call from inside the energy_full() function before it returns.
The original write_restart function (Output::write_restart) is modified such that it writes replaces the aestrik ‘*’ in filenames with the argument it receives (instead of the ntimestep). This is used in combination with a variable (energy_calls) to count the number of energy_full() was calls was used to get distinct restart files for each energy_full() calls.
This method was working very well for cases with Pure LJ and for LJ with P3M (1e-5 precision).
1.2 GCMC Energy
Apart from getting the system state, fix calls were added inside FixGCMC::energy_full() was changed such that it would resemble Verlet::run() with fix elecrode/conp. Table in the trials section lists all the additional modify functions that were added to the FixGCMC::energy_full() corresponding to a version index (s, se and senp).
- The s version corresponds to the original FixGCMC::energy_full().
- The se version includes all the functions from s and additionally modify functions that are part of the fix_electrode_conp mask (pre_exchange [modified, see below], post_neighbor, pre_reverse. pre_force exists in FixGCMC::ebergy_full() by default).
- The senp version has all the modify member calls as in the se version except for the modifed (see below) pre_exchange call.
Since, FixGCMC in itself is listed in the list_pre_exchange fix list, a modified pre_exchange_exclude function was added to modify.cpp (and modify.h) which accepts the id of the calling fix as an argument and executes all the fixes inside list_pre_exchange except the calling/invoking fix.
Lastly. utils::logmesg is used to print the energy from FixGCMC::energy_full() before the function returns.
2. Trials
I have used an LJ tube with a single charged particle as my initial system for simulation. The force field is a hybrid one combining mie/cut and coul/long. PBC is assumed along all the axes making the tube infinite along z.
I request you to find the rest of my simulation files in the 1.mie_pppm_electrode folder (shared previously).
Below is a table of energies that I obtain from the simulation based on my FixGCMC::energy_full() version corresponding to the function calls. The Y in table represnet presence of the modify call and N represent absence. The force_clear() in GCMC is implemented differently from that of verlet (and hence marked Y*). The energy provided for gcmc (second to last row) corresponds to the restart index 3 (elec_mc_3.restart in 1.mie_pppm_electrode folder/1.*.mc_* folders here).
| modify calls | s | se | senp |
|---|---|---|---|
| post_int | N | N | N |
| pre_exchg | N | Y | N |
| comm_exchg | Y | Y | Y |
| pre_neigh | Y | Y | Y |
| post_neigh | N | Y | Y |
| frc_clr | Y* | Y* | Y* |
| pre_force | Y | Y | Y |
| pre_rev | N | Y | Y |
| comm_rev | N | Y | Y |
| post_force | Y | Y | Y |
| eof_step | N | N | N |
| energies | s | se | senp |
| gcmc | 327333560.79 | 327337203.67 | 327337203.67 |
| md_restart | 148.807581 | 148.807581 | 148.807581 |
Note: Only the algo cg (1e-20) option of the electrode/conp works when used in combination with GCMC. Both the other algos (mat_inv and mat_cg) ends up giving me segmentation faults upon running.
There is large energy differences between the GCMC and the MD verlet here obtained from restart.
Also, the wall charges from the GCMC do not correspond to that obtained from MD when run from the restart.
Question
As mentioned, I would like to ensure that the electrode runs with GCMC reproducing energies consistent with a normal verlet run. It would be helpful if someone can throw some more insight into why I am not able to reproduce the energies properly. I can also share you any other data related to the case if needed.
EDIT-1 (13/4/24)
Below is an image for clarity !! I want to ensure (energy gcmc) e_gcmc = e_lammps-md