Thank you for your reply.
I tried to achieve the same (hard particle in an alloy matrix) by using in.shear example from lammps examples and it is able to generate dislocations well. I wanted to calculate the shear stress at the same time so I am using compute stress atoms to get the stress in the mobile region. I have marked it in bold in my code below.
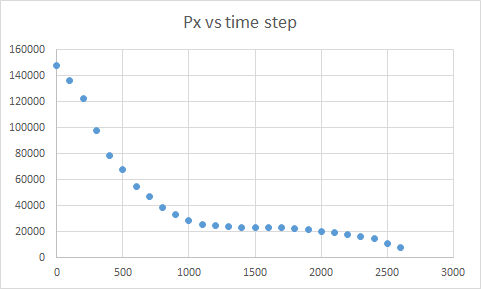
The problem is that I see a decreasing shear stress. I would expect an increasing trend since we are shearing the box and the stress should rise with strain.
Could you kindly tell me where I am going wrong?
Also what would be an ideal approach to calculate the shear strain in this example?
units metal
boundary s s p
atom_style atomic
lattice fcc 3.52
region box block 0 16.0 0 10.0 0 8 #2.828427
create_box 7 box
lattice fcc 3.52 orient x 1 0 0 orient y 0 1 1 orient z 0 -1 1 &
origin 0.5 0 0
create_atoms 1 box
variable fa equal 50000
variable fb equal 37609
variable fc equal 24933
variable fd equal 12421
variable fe equal 10
variable ff equal 1
variable ft equal 62500
set type 1 type/fraction 2 (v_fa/v_ft) 1734536
set type 2 type/fraction 3 (v_fb/(50000)) 1734535
set type 3 type/fraction 4 (v_fc/(37609)) 1734534
set type 4 type/fraction 5 (v_fd/(24933)) 1734533
set type 5 type/fraction 6 (v_fe/(12421)) 1734533
set type 6 type/fraction 7 (v_fe/(12457)) 1734533
group Co type 1
group Cu type 2
group Cr type 3
group Fe type 4
group Ni type 5
group W type 6
group Al type 7
pair_style eam/alloy
pair_coeff * * CoCuCrFeNiWAl.set Co Cu Cr Fe Ni W Al
neighbor 0.3 bin
neigh_modify delay 5
region lower block INF INF INF 0.9 INF INF
region upper block INF INF 6.1 INF INF INF
group lower region lower
group upper region upper
group boundary union lower upper
group mobile subtract all boundary
set group lower type 2
set group upper type 3
void
region void1 sphere 8 3.5 3 2 side in
create_atoms 1 region void1
group void1 region void1
set group void1 type 7
region void2 sphere 8 3.5 3 1 side in
#lattice bcc 3.17 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
create_atoms 1 region void2
group void2 region void2
set group void2 type 6
fix 111 void2 rigid single
neighbor 2.0 bin
neigh_modify delay 10 check yes